
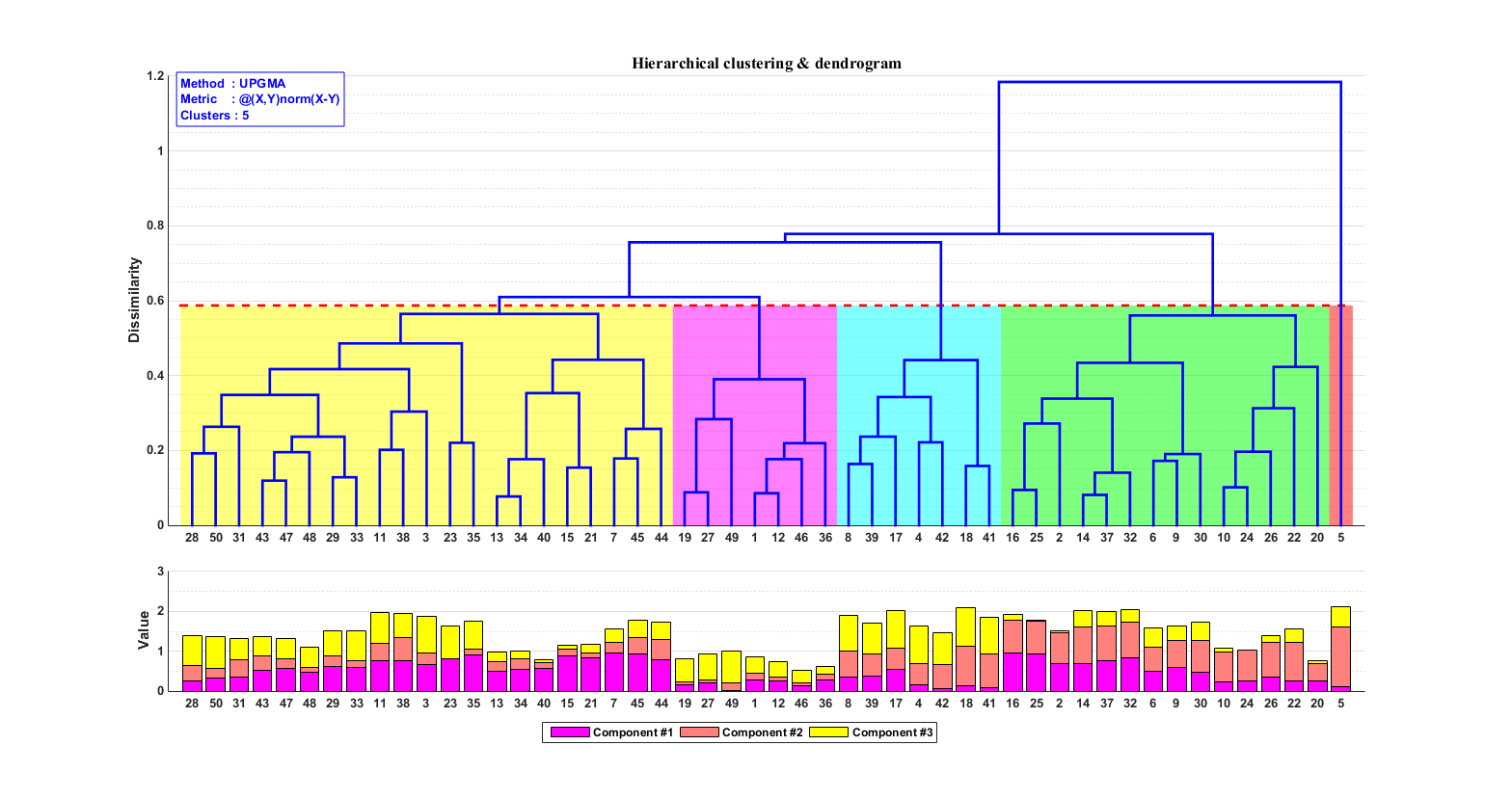
估计阅读时长: 14 分钟https://github.com/xieguigang/sciBASIC 层次聚类通过计算不同类别数据点间的相似度来创建一棵有层次的嵌套聚类树。基于层次聚类分析,我们可以初步可视化我们的一些原始数据: 例如对样本的层次聚类分类,可以让我们了解到样本在分组之间以及分组内的异质性。 对生物序列进行基于相似度的层次聚类分析,我们可以了解到序列之间的相似性程度或者进化关系 Order by Date Name Attachments metabolome • 14 kB • 700 click […]
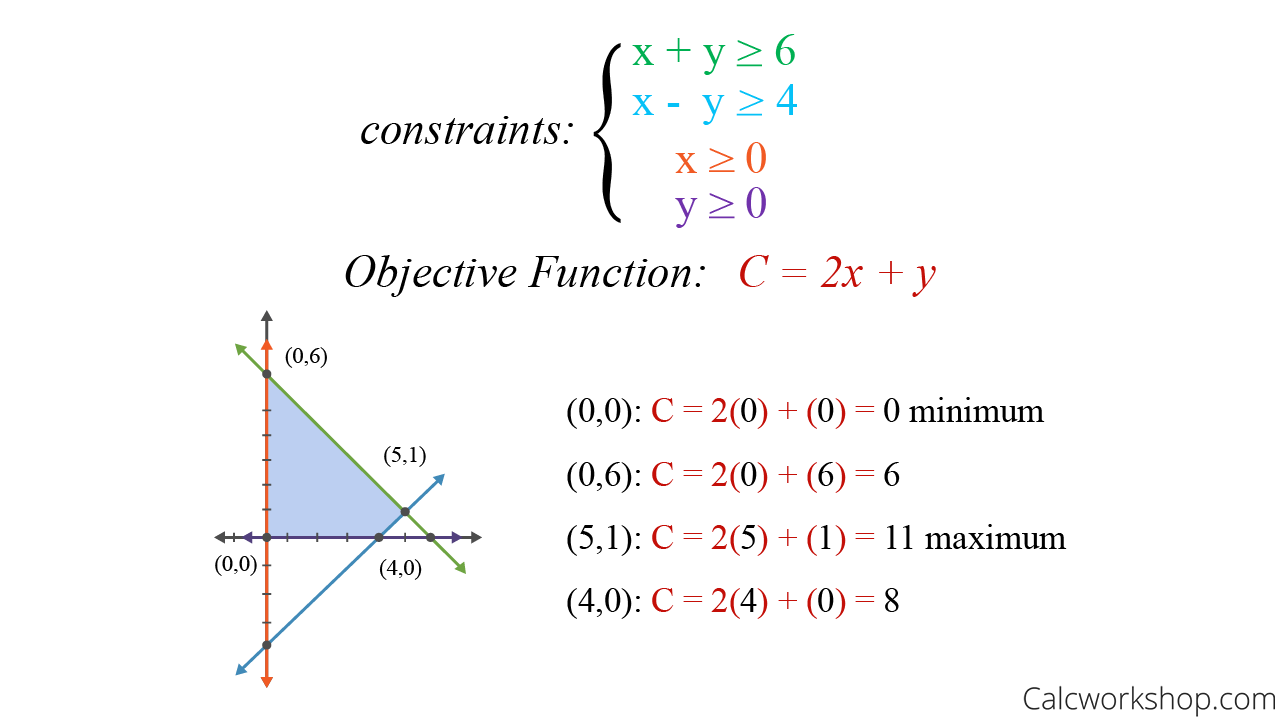
估计阅读时长: 30 分钟https://github.com/xieguigang/sciBASIC/ 线性规划(Linear programming,简称LP)方法起源于20世纪40年代,由美国数学家乔治·丹齐格(George Dantzig)提出,并设计了著名的“单纯形法”。这种优化算法是运筹学中研究较早、发展较快、应用广泛、方法较成熟的一个重要分支,它是辅助人们进行科学管理的一种数学方法。研究线性约束条件下线性目标函数的极值问题的数学理论和方法。通俗点的来讲,就是我们基于这一种数学优化技术,用于在一组线性约束条件下,求解线性目标函数的最大值或最小值(就是在“有限资源”和“一定规则”下,找到“最佳方案”的一种方法)。 Order by Date Name Attachments linear-programming-example • 22 kB • 770 click […]
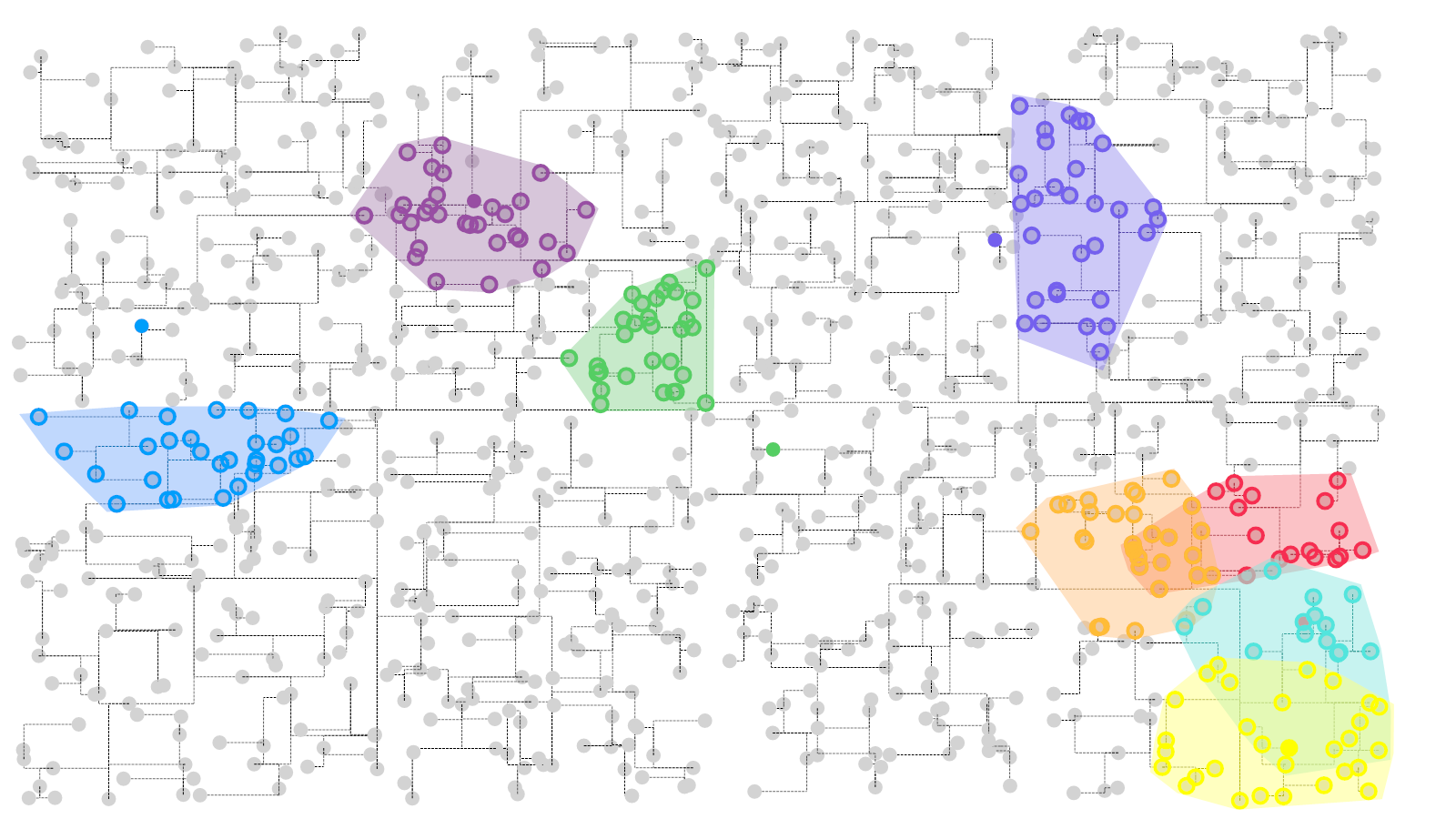
估计阅读时长: 8 分钟https://github.com/xieguigang/sciBASIC 在进行无监督聚类分析的方法之中,我们在算法代码之中一般会遇到求解与某一个样本数据点最相似的数据点的计算过程。对于这个计算过程,一般而言我们是基于欧几里得距离来完成的。 Order by Date Name Attachments Visual a KDtree Search • 274 kB • 750 […]
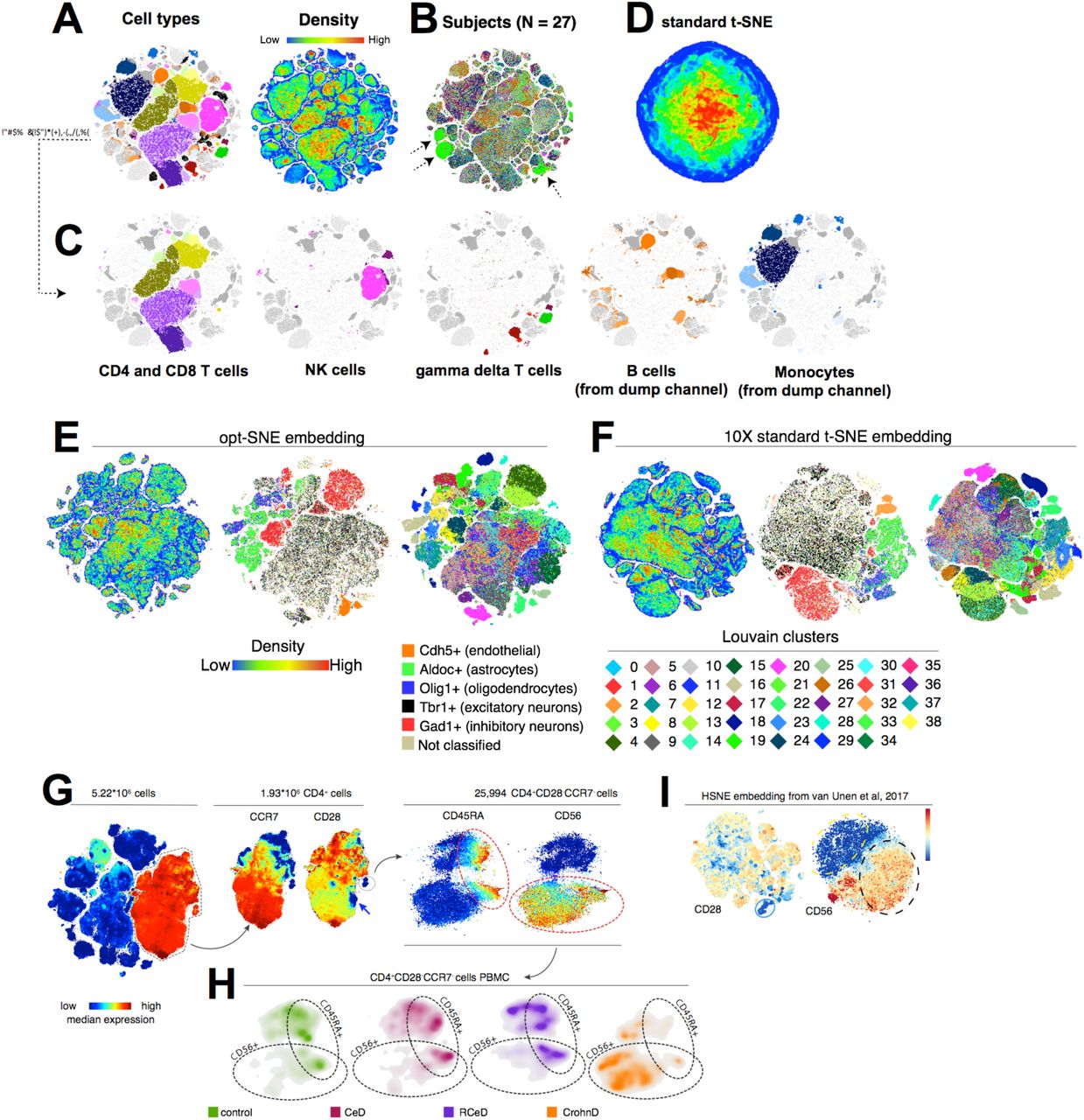
估计阅读时长: 11 分钟PhenoGraph提供了与UMAP类似的算法过程进行单细胞组学数据的细胞分型处理操作。与UMAP方法相比,PhenoGraph并不会产生数据降维效果,仅仅产生数据点Cluster信息。如果需要将数据进行可视化,还需要借助于t-SNE算法将PhenoGraph的分型结果数据投影到一个二维平面上完成。 Order by Date Name Attachments Phenograph-image4 • 200 kB • 689 click 2021年8月9日Automated Optimal Parameters […]

估计阅读时长: 11 分钟https://github.com/xieguigang/sciBASIC Louvain算法是基于模块度的网络节点集群发现算法。该算法在效率和效果上都表现较好,并且能够发现层次性的网络节点集群结构,其优化目标是最大化整个网络集群模块的模块度(Modularity)。 Order by Date Name Attachments graph • 2 MB • 707 click 2021年8月7日Metavirome network […]
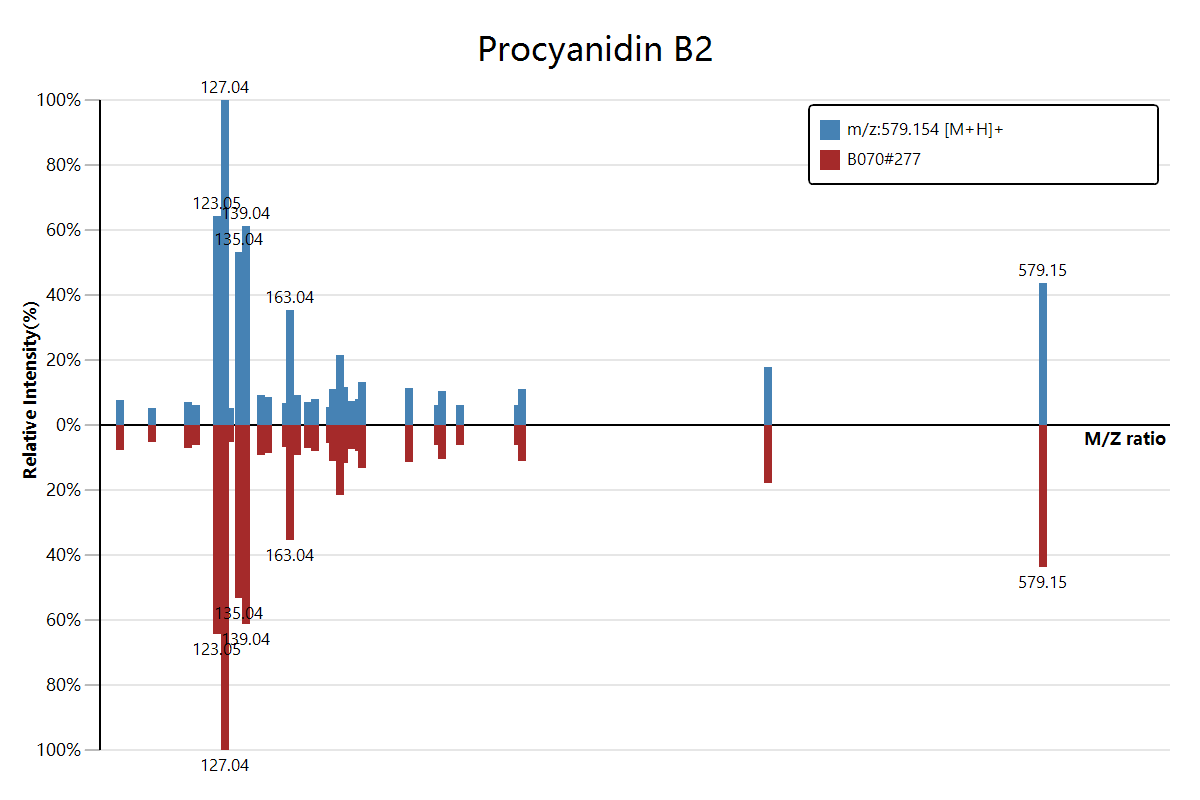
估计阅读时长: 2 分钟https://mzkit.org/ 代谢组文章整个坎坷的发表经历中,大家可能都会遇到的一个老大难问题就是我们有时候会需要从原始数据中得到物质注释结果的二级质谱图数据。对于熟悉xcms程序包的同学,获取二级质谱图可能会比较容易:无非就是加载原始数据,然后按照m/z和rt找到对应的二级scan就好了。但是,这种方法会需要编写脚本来完成。 Order by Date Name Attachments download • 33 kB • 700 click 2021年8月4日[278][MS_MS] FTMS […]

估计阅读时长: 2 分钟https://github.com/dotvanilla/vanilla 在Vanilla编译器项目之中,会需要一个程序模块将VisualBasic代码进行解析为语法树。然后我们基于此语法树就可以将VisualBasic项目转换为WAST源代码,从而实现编译为WebAssembly程序了。在这个步骤之中,我们可以通过一个微软官方的Roslyn编译器平台来实现。 Order by Date Name Attachments Roslyn-nuget • 107 kB • 680 click 2021年7月24日what-is-visual-studio • […]
估计阅读时长: < 1 分钟https://github.com/xieguigang/codegraph Attachments Microsoft.VisualBasic.Framework_v47_dotnet_8da45dcd8060cc9a.dll • 10 MB • 601 click 2021年8月29日
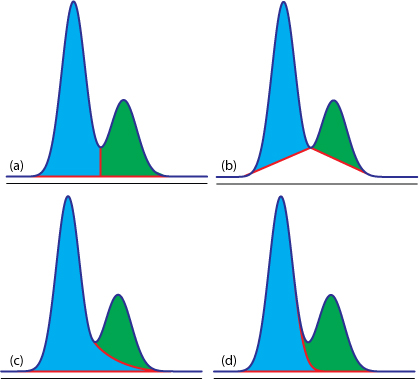
估计阅读时长: 8 分钟https://github.com/xieguigang/sciBASIC/tree/master/Data_science/Mathematica/SignalProcessing 进行峰识别是在代谢组学原始数据分析之中进行定量分析的很重要的一环。在代谢组学之中,定量分析分为靶向定量,以及非靶向定量计算这两大部分。 Order by Date Name Attachments Figure12.36 • 50 kB • 718 click 2021年7月10日view_signal • […]

估计阅读时长: 9 分钟https://github.com/dotvanilla/vanilla WebAssembly是一种运行在浏览器端的二进制程序集文件。和普通的应用程序开发一样,WebAssembly需要基于一定的源代码文本进行编译。这个编译所需要的源代码文本就是WAST文件。 Understanding WebAssembly text format(https://developer.mozilla.org/en-US/docs/WebAssembly/Understanding_the_text_format)

[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?
谢老师,写快点呀,在看着你更新文章呢。
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]