
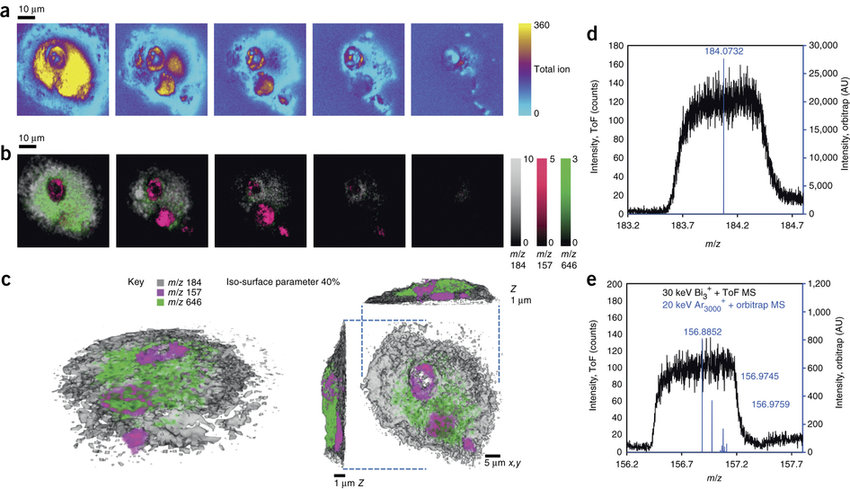
估计阅读时长: 2 分钟在BILIBILI上观看视频:【空间代谢组学】AP-MALDI 质谱成像技术介绍 哈啰,大家好呀,鸽了大半年之后,你们的小姐姐又回来啦。为了更好的制作出质量更高的视频,你们的六神无主鸠小姐姐呀,在这大半年的时间里面一直在努力的学习新技术。经过半年的钻研学习,收获满满。谈到最近几年的热门尖端技术,大家都会谈论到空间转录组和单细胞技术。一般而言,代谢组学的发展要稍微滞后于转录组学研究。最近一年呢,随着空间转录组的热度的降低,空间代谢组的热潮也终于姗姗来迟终于到来了。今天呢,我想要为大家介绍的是在最近几年内出现的,目前比较火热的空间代谢组学研究领域内的质谱成像技术。 Order by Date Name Attachments 3D-MS-imaging-using-dual-beam-and-dual-spectrometer-mode-10-of-single-rat-alveolar • 99 kB • 790 click 2022年5月6日Microsoft […]
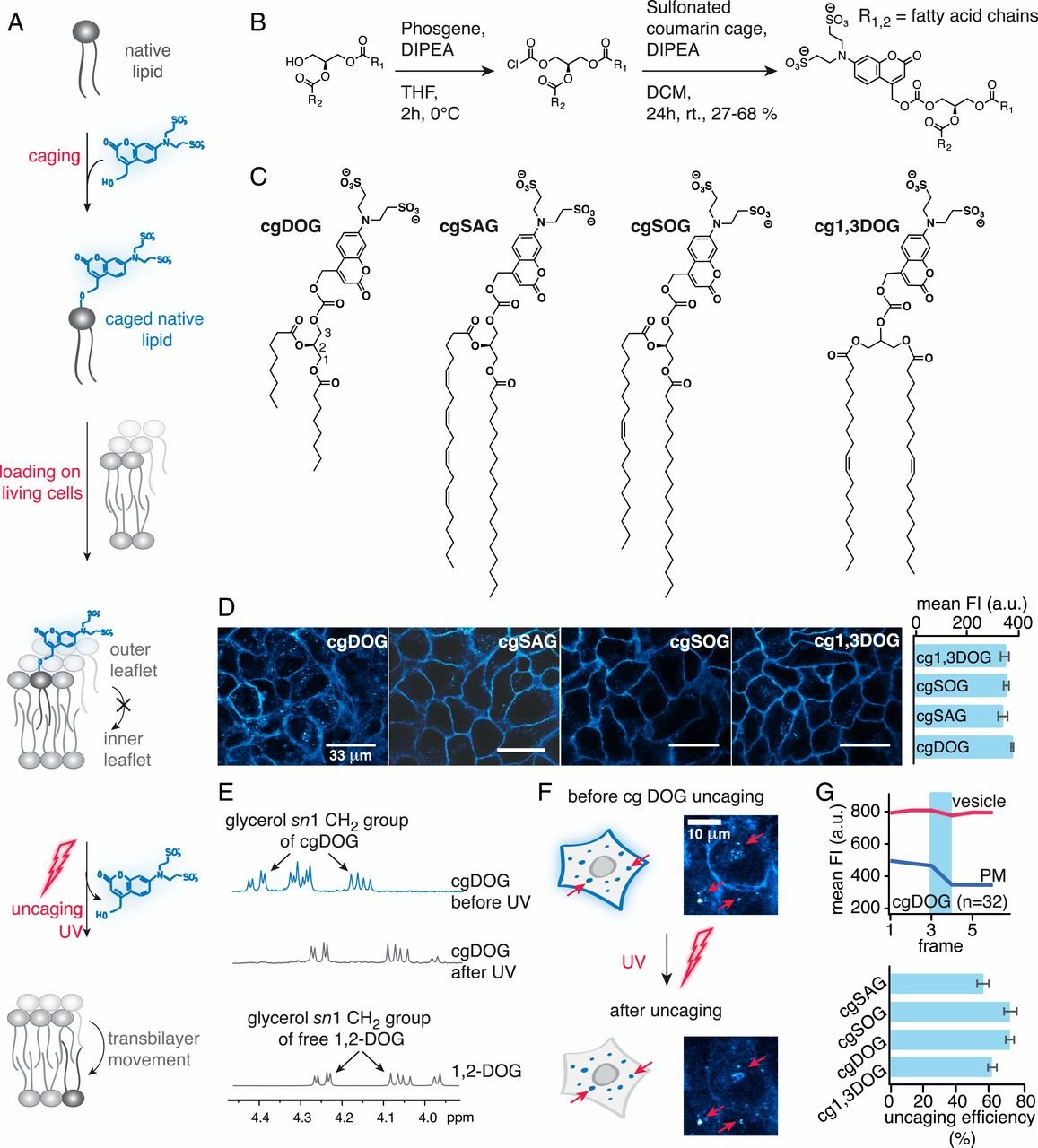
估计阅读时长: 17 分钟脂质的分类 LipidMAPS是美国国立卫生院推进的“脂质代谢途径研究计划”,涵盖目前最权威的脂类分类、命名法和结构信息,还囊括了众多的脂质组定性定量方法。此外,它还提供了一些生物信息学分析工具,比如基于质谱的脂质定性工具,通过给定特定的m/z,或特征子离子信息,或二级谱图信息,可以预测可能的脂质分子;又比如可以管理、可视化及编辑脂质信号通路等。 Order by Date Name Attachments gr1_lrg • 379 kB • 713 click 2022年4月30日MS-based lipidomics […]
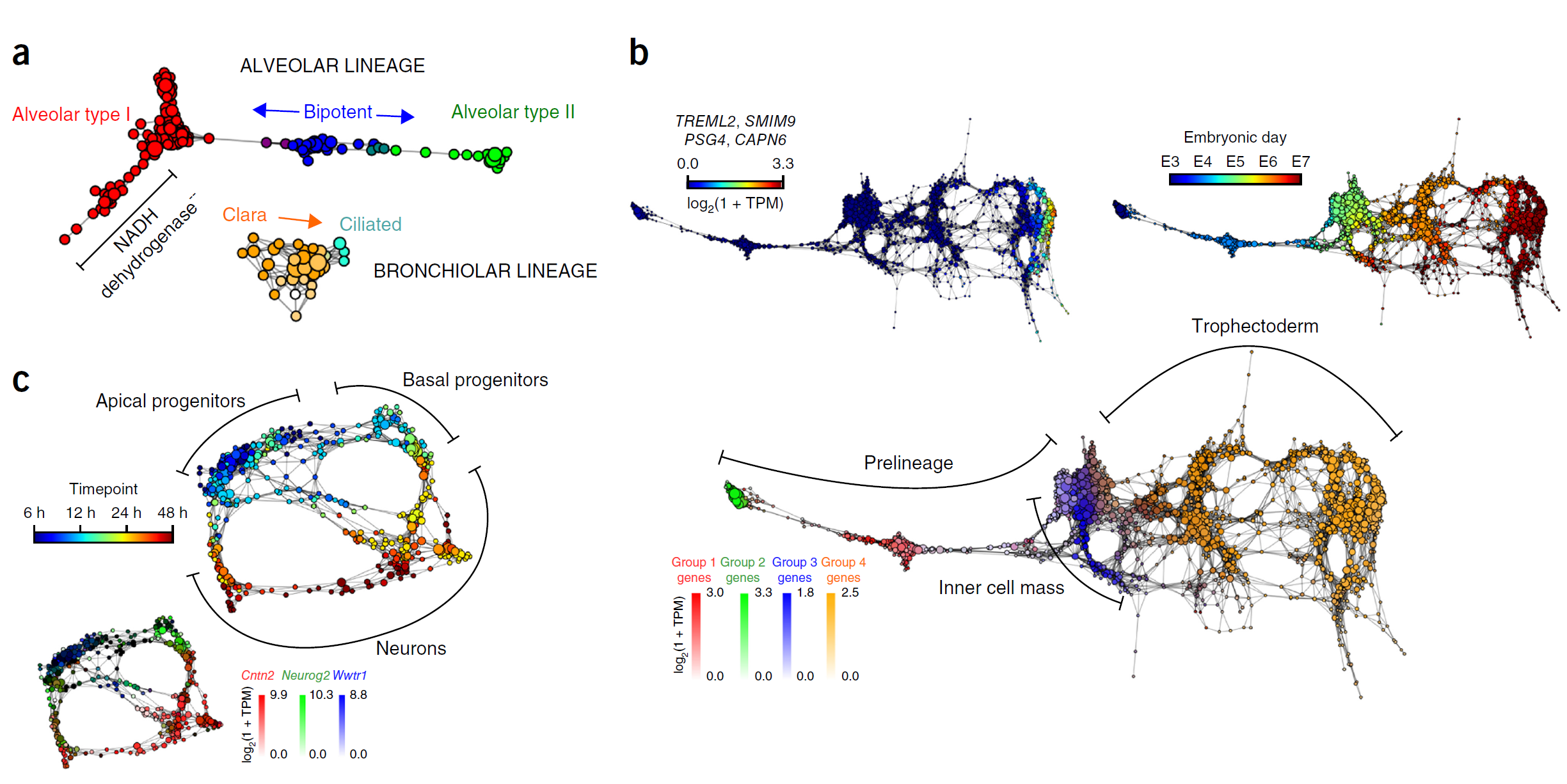
估计阅读时长: 14 分钟单细胞分析方法学习文献打卡记录: 【单细胞组学】PhenoGraph单细胞分型 【单细胞分析方法】VeTra:基于RNA速度的轨迹推断工具 【单细胞分析方法】单细胞图嵌入 Order by Date Name Attachments Cellular populations during motor neuron differentiation • […]
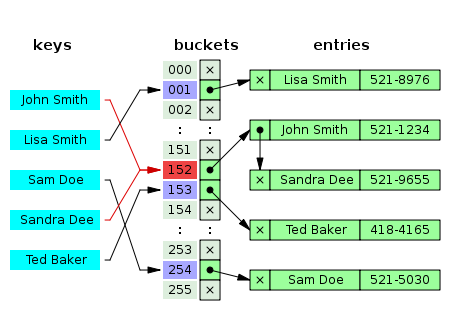
估计阅读时长: 8 分钟在之前的BioDeep代谢物数据库整合工作之中,所提取的代谢物注释信息的唯一编码是来自于数据库表之中的递增主键。由于数据库之中的递增主键的唯一编码值是与数据内容完全无关的数据,所以在基于图数据库做数据库整合的结果在两次整合操作之后,可能会因为先后输出顺序不一致的原因,得到的在关系型数据库中的唯一递增编号可能会完全不一样了。这个问题会对数据库更新操作造成非常大的困扰。 Order by Date Name Attachments 450px-Hash_table_5_0_1_1_1_1_1_LL • 26 kB • 636 click 2022年4月16日metadata-table • 58 […]
估计阅读时长: 7 分钟Assembles a manifold that is defined through a series of overlapping, locally-defined PCA subspaces. Non-mutual k-nearest-neighborhoods […]
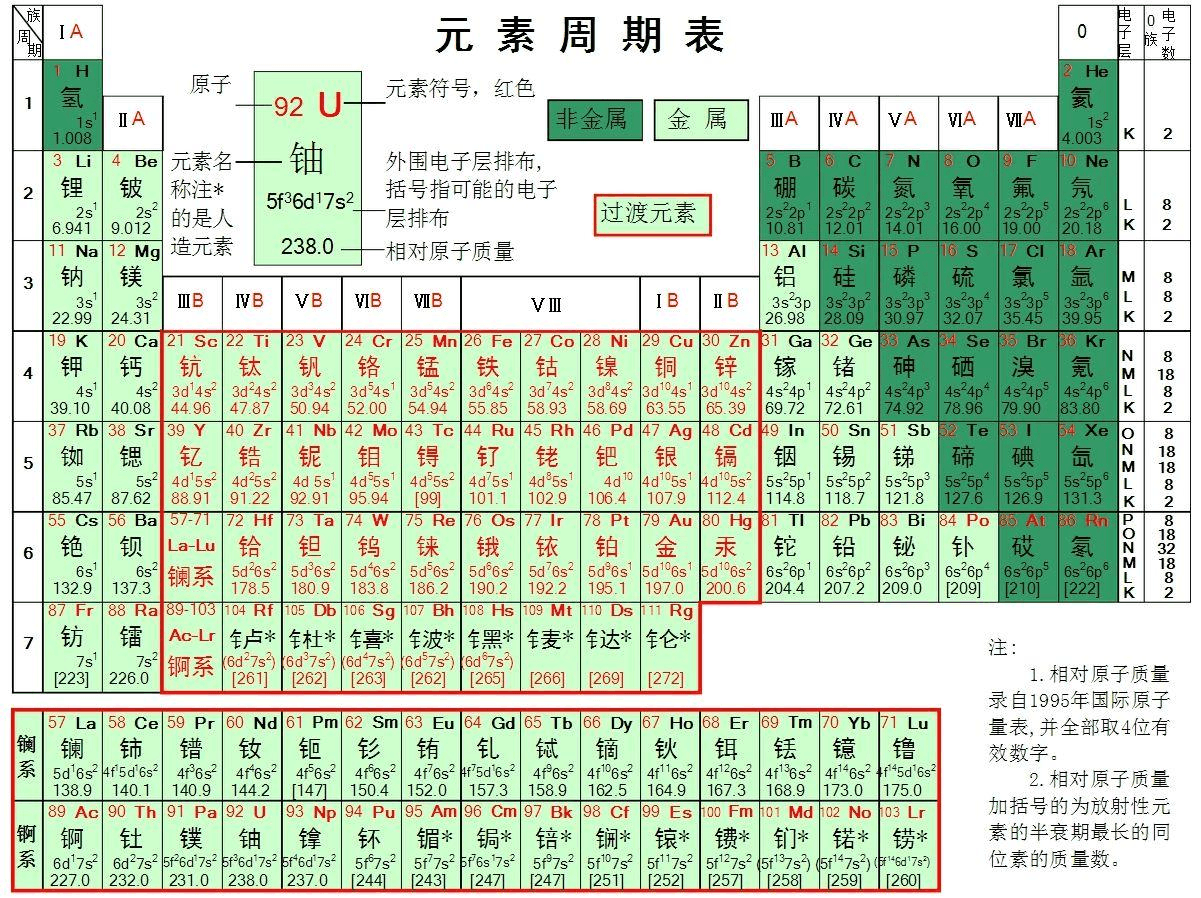
估计阅读时长: 5 分钟目前我们根据质谱数据进行代谢物ROI注释分析,很大一部分的工作是建立在已经可以被纯化的化合物的纯标准品所建立的标准品库数据的比对操作之上的。但是依赖于质谱参考谱图数据库所完成的代谢物注释分析,也仅能够得到很小的一部分结果,因为能够纯化或者合成的化合物在整个自然界中目前只占比较小的一部分。并且购买标准品也会需要耗费大量的实验室资金预算。 Order by Date Name Attachments The-Periodic-Table • 2 MB • 716 click 2022年3月20日Leucine[M+H]+ • 33 […]
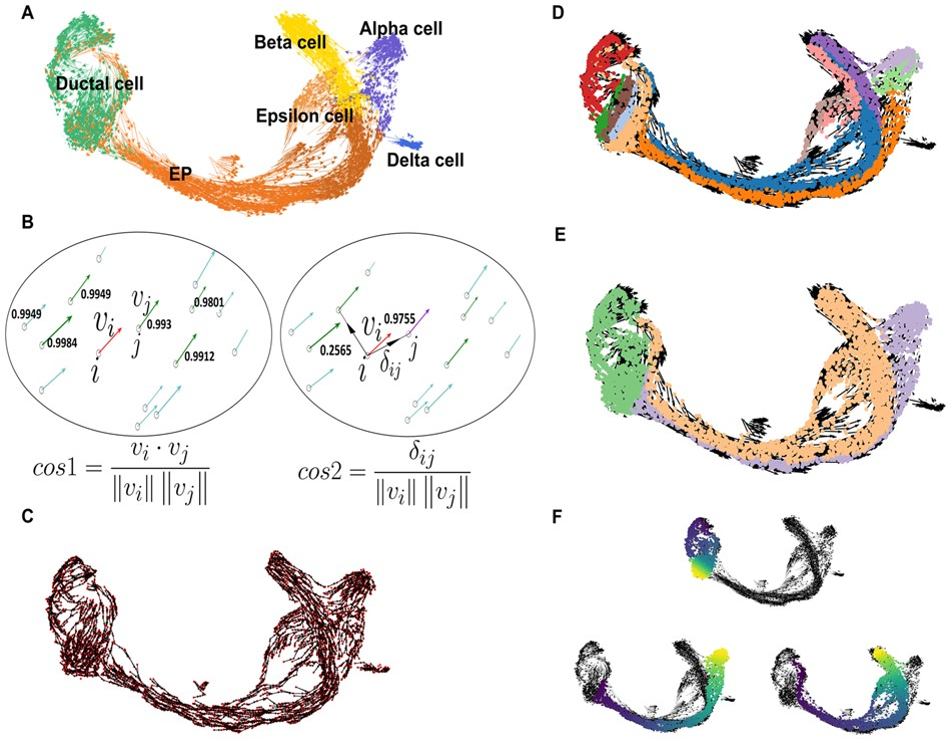
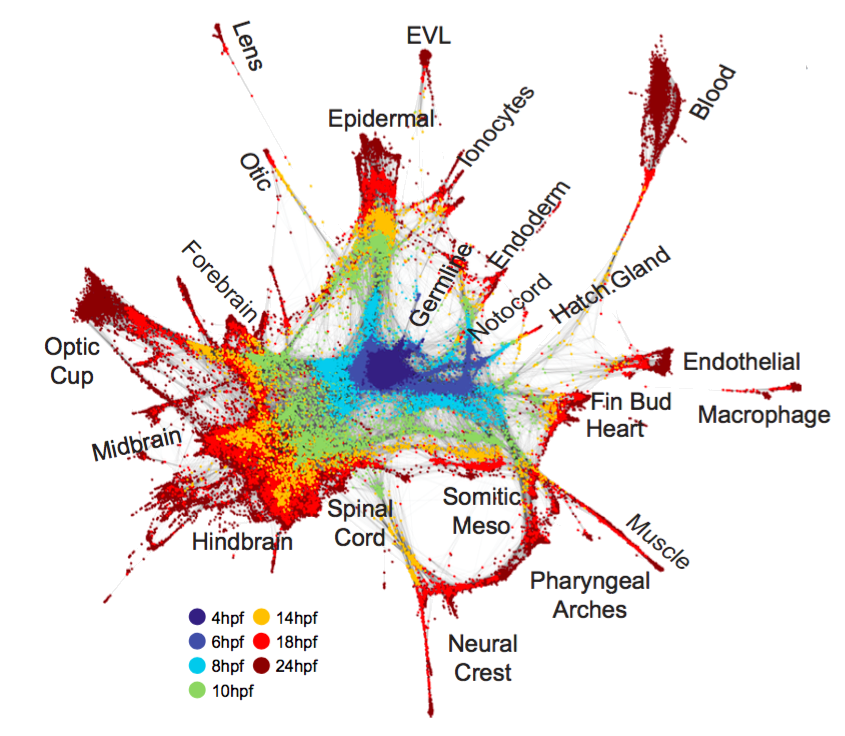
估计阅读时长: 5 分钟单细胞轨迹可以揭示基因调控如何控制细胞命运:大多数细胞状态转变,无论是在发育,重编程或者是疾病异常状态,都以基因表达变化的级联为特征。 Order by Date Name Attachments vec • 722 kB • 680 click 2022年3月17日Slide10 • 14 […]

估计阅读时长: 5 分钟https://github.com/xieguigang/graphQL 构建一个图数据库,可以用来帮我们解决复杂的知识关联计算问题。例如我们想要程序向我们回答dihydrogen oxide与water是否是同一个东西。如果光从字符串比较角度上面来看待这个问题的话,很显然,二者的字符串比较结果肯定是False。面对上面的这个问题,图数据库则可以很简单的向我们回答道上面的两个字符串都是指代的同一个东西。 Order by Date Name Attachments tumblr_inline_mqvdlydGCp1qz4rgp • 124 kB • 621 click 2022年3月5日Capture […]
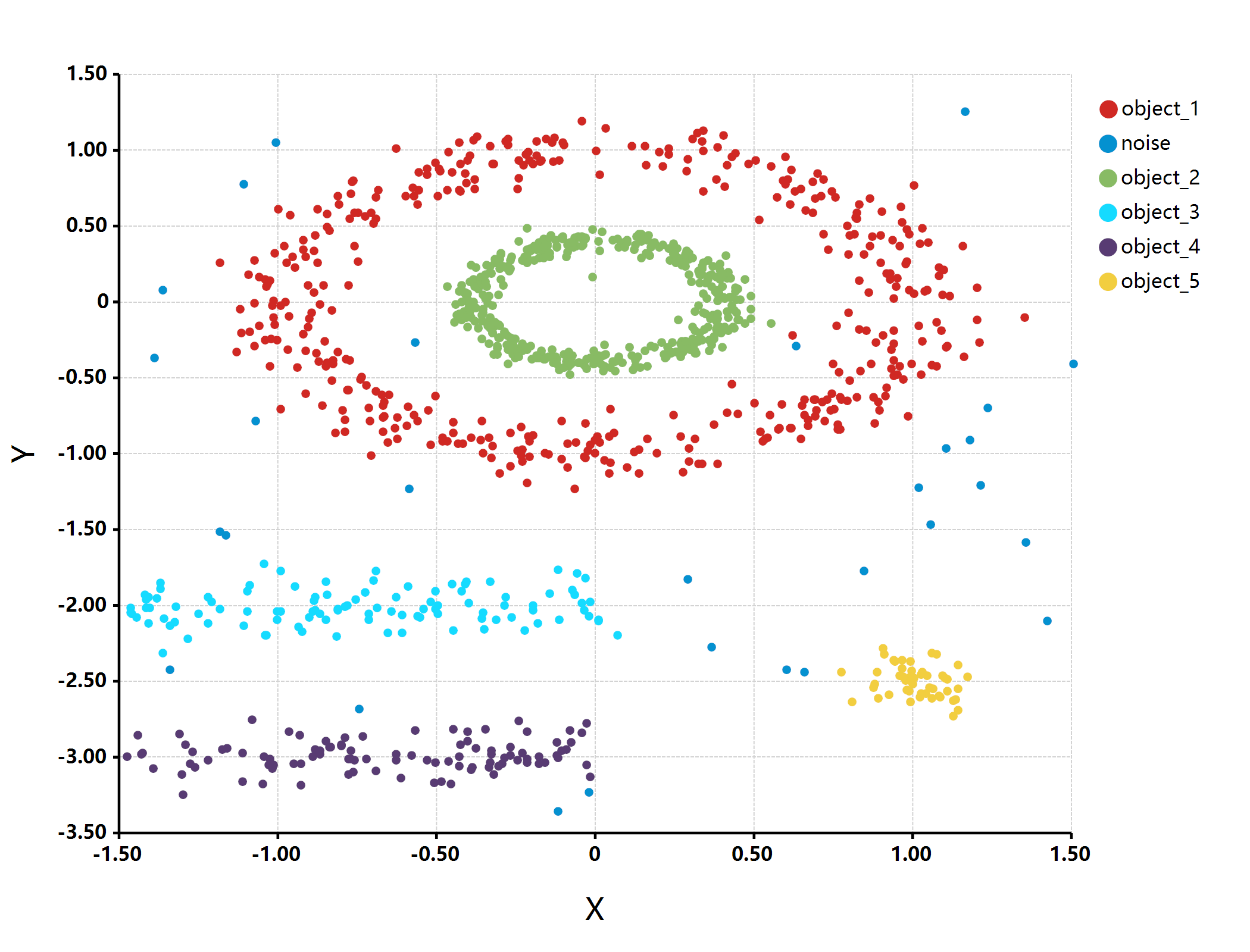
估计阅读时长: 2 分钟imports "clustering" from "MLkit"; require(graphics2D); multishapes = read.csv("./multishapes.csv"); [x, y] = list(multishapes[, "x"], multishapes[, "y"]); print(multishapes, […]

估计阅读时长: < 1 分钟https://github.com/rsharp-lang/R-sharp 前言 经过了2021年一年时间的奋斗,目前R#脚本语言环境终于可以算是能够支撑起比较完整的数据分析流程了。在2021年这段时间,我为R#脚本语言环境大概做了以下几件我认为是比较里程碑式的工作: 建立起了一个比较成熟的脚本打包系统 仿照R语言引入了ggplot和ggraph类似的作图系统 借助于mzkit的开发,将R#语言成功的应用于商业化的质谱数据分析产品之中 为了扩大R#语言环境的受众,在2022年初,也就是这个月内,我相继为Python语言和Julia语言添加了对R#语言环境的支持。下面我们就来聊聊在R#语言环境中的对上面所提到的两种语言的支持。 Order by Date Name Attachments programming • 262 kB […]

[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?
谢老师,写快点呀,在看着你更新文章呢。
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]