
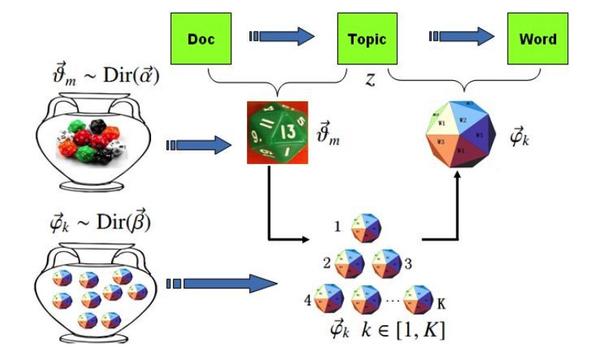
估计阅读时长: < 1 分钟LDA(Latent Dirichlet Allocation,潜在狄利克雷分配)是一种主题模型,它假设文档是由多个主题混合而成的,而每个主题又是通过一定的概率分布选择词语生成的。Gibbs LDA 的核心在于使用吉布斯采样方法来推断这些隐藏的主题分布。 Attachments v2-883ac9db7f1cbd7325b2450cd225a897_b • 29 kB • 13 click 2026年2月23日
估计阅读时长: 17 分钟EC Number是国际酶学委员会(IUBMB)制定的一套酶分类编号体系,EC Number采用层级分类法,由4个数字组成,分别代表酶的大类、亚类、亚亚类和序号。例如,“EC 1.1.1.37”中,第一个“1”表示氧化还原酶大类;第二个“1”表示作用于CH-OH基团;第三个“1”表示以NAD+或NADP+为受体的酶;第四个“37”表示特定酶苹果酸脱氢酶。这种层次结构意味着EC编号蕴含了丰富的功能信息,包括酶催化的反应类型和底物/机制。将EC Number嵌入为向量,有助于我们利用机器学习模型进行功能预测、相似性分析等。 Order by Date Name Attachments Capture • 14 kB • 105 […]
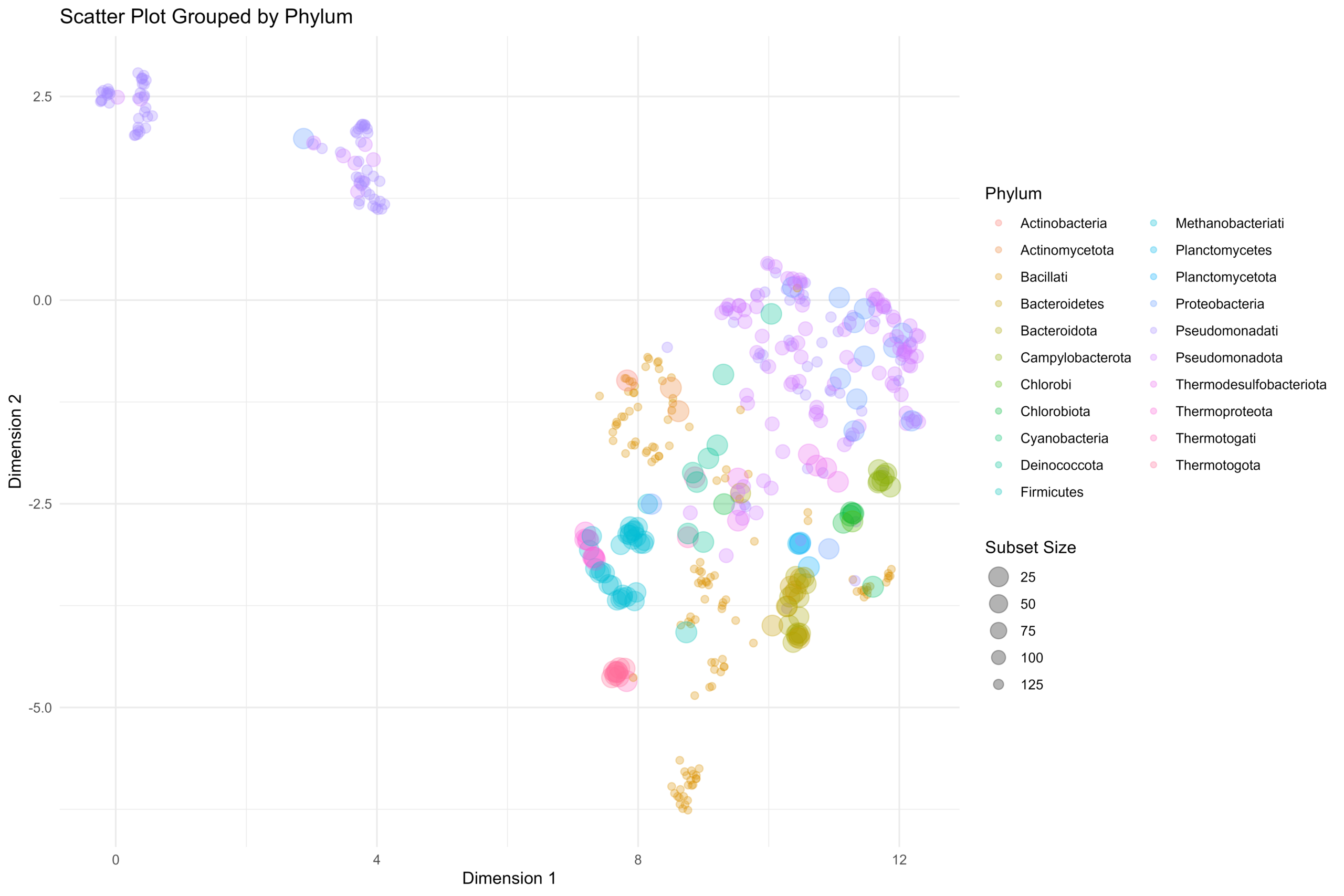
估计阅读时长: 11 分钟在将生物序列(如基因组或蛋白质序列)或文本数据转换为数值向量形式时,TF-IDF(Term Frequency-Inverse Document Frequency)和N-gram One-hot(又称Bag-of-n-grams)是两种经典且基础的文档嵌入算法。它们各自侧重于不同的特征提取方式,常被用于自然语言处理和生物信息学领域。 Attachments scatter_plot • 433 kB • 108 click 2026年2月10日
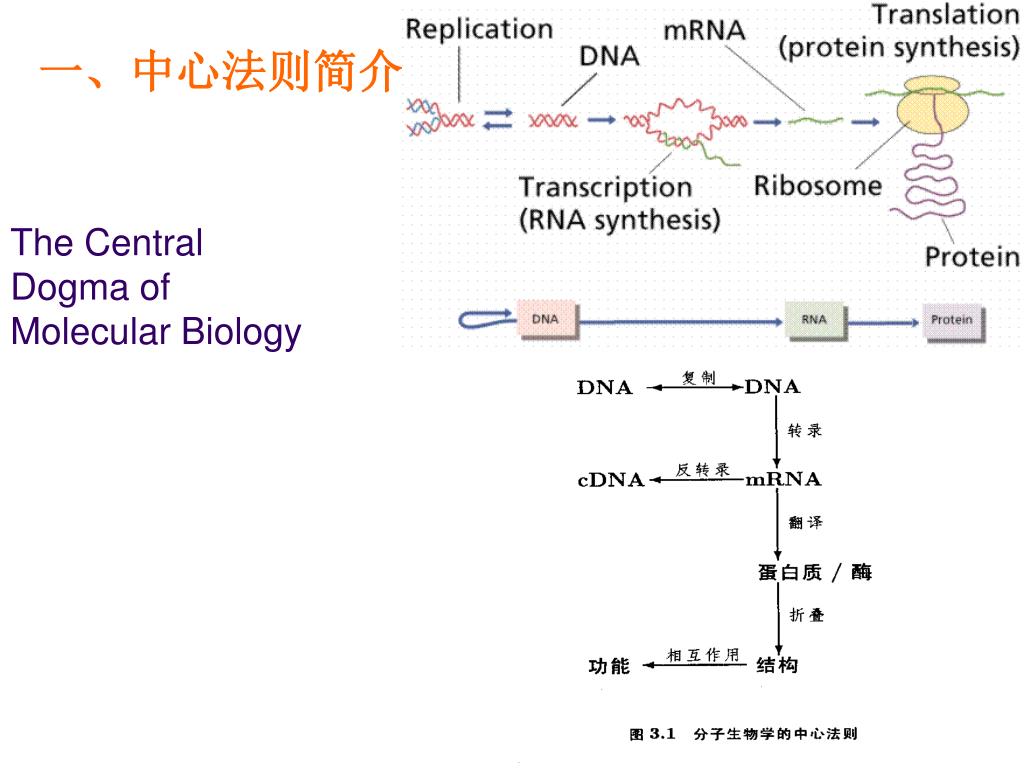
估计阅读时长: 8 分钟原核生物细胞内的中心法则是指遗传信息从DNA经RNA到蛋白质的传递过程,具有高效和经济的特点。DNA复制、转录和翻译均在细胞质中进行,且转录与翻译高度偶联——新生mRNA尚未完全合成,核糖体便已结合并开始翻译,极大提升了蛋白质合成速率。原核生物mRNA常为多顺反子结构,一条mRNA可编码多个功能相关的蛋白质,且无内含子、无需剪接,可直接作为翻译模板。此外,原核mRNA半衰期极短,便于快速响应环境变化。基因表达主要通过操纵子结构在转录水平进行精细调控,如乳糖操纵子和色氨酸操纵子,使原核生物能够灵活适应多变环境。这些机制共同构成了原核生物中心法则的核心,体现了其高度优化的遗传信息传递系统。 Attachments the-central-dogma-of-molecular-biology1-l • 70 kB • 225 click 2025年12月21日
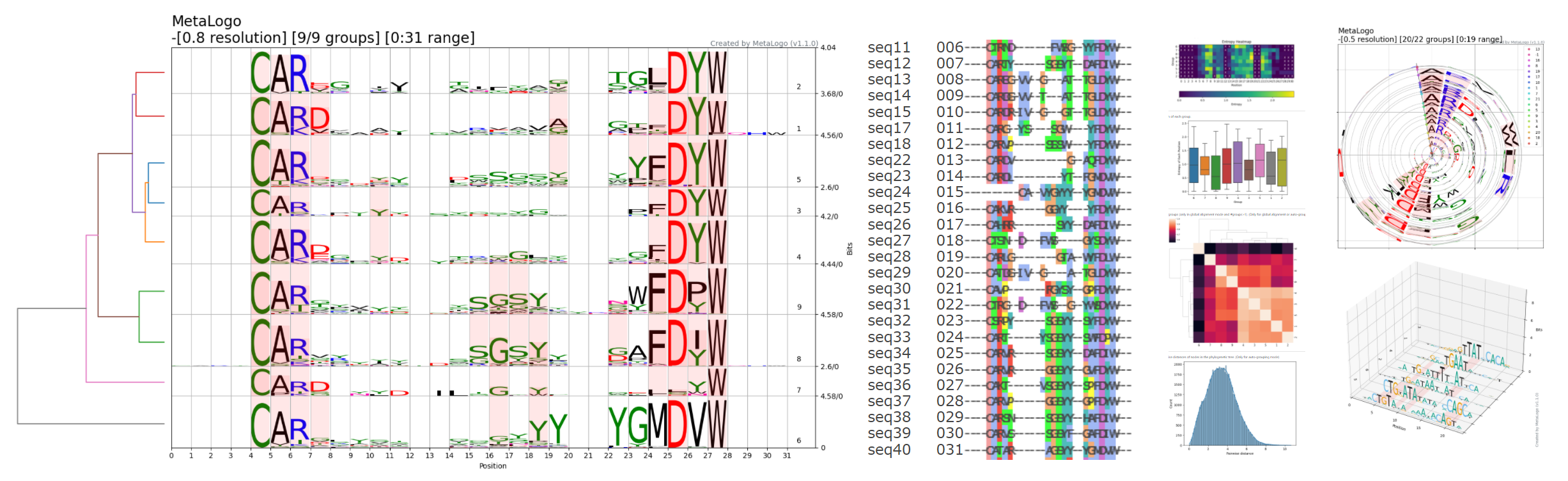
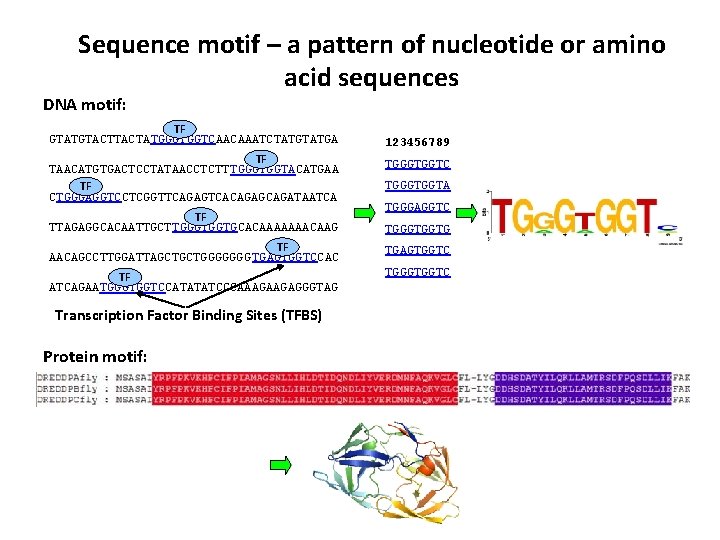
估计阅读时长: 23 分钟Sequence Logo 是一种可视化 DNA 或蛋白质序列保守性的图形表示方法。每个位置(列)上的字母堆叠高度代表该位点的信息含量(以 bits 为单位),而每个字母的高度则与其在该位点出现的频率成正比。高信息量的位置字母堆得高,低信息量的位置则矮甚至接近零。Sequence Logo的绘制遵循信息熵原理,我们可以很直观的通过某一个位置的总高低来了解该处位置的信息含量有多少,高信息量的位置,字母堆的高,一般会出现某一个字符特别高,表明该处非常保守。 位置权重矩阵(Position Weight Matrix, PWM)是描述基因组调控因子结合位点序列模式的核心模型。它通过统计在结合位点序列中每个位置上各核苷酸(或氨基酸)出现的频率,来量化该位置对不同碱基的偏好程度。PWM通常以矩阵形式表示,行对应核苷酸(A、C、G、T/U),列对应序列中的位置,矩阵元素即为该位置该核苷酸相对于背景的权重得分。这一模型简洁且易于计算,因此在转录因子结合位点(TFBS)等调控元件的识别和表征中被广泛采用。 Order by Date Name […]

估计阅读时长: 5 分钟将复杂的生物学过程拆解为单元化学反应,是进行定量模拟的基石。转录是基因表达调控的关键环节,决定了细胞在特定时间、特定环境下合成哪些蛋白质,对生命活动至关重要。最近的工作中需要将原本非常粗糙的虚拟细胞转录事件模型拆解为更加细分化的多步骤生物化学过程,以适应针对细胞群落生长的建模计算。下面为我将原核生物的转录过程拆解为一系列可以用化学式表示的单元步骤的结果。 在介绍这些分步骤之前,我们会需要首先来定义一下模型中会用到的各种“化学物质”(分子和复合物): RNAP: RNA聚合酶全酶(包含核心酶和σ因子)。 DNA: 基因组DNA双链。 DNA_P: 包含启动子区域的DNA。 DNA_T: 包含终止子区域的DNA。 NTP: 核糖核苷三磷酸(ATP, UTP, GTP, CTP的统称)。 PPi: […]
估计阅读时长: 30 分钟零分布(null distribution)是指在假设零假设(null hypothesis)成立的情况下,某个统计量随机取值的概率分布。在统计假设检验中,我们通常提出一个零假设(例如“两组数据没有显著差异”或“观察到的模式仅由随机因素造成”),然后根据观测数据计算一个检验统计量。零分布描述了这个统计量在零假设为真时的分布情况。通过将实际观测到的统计量与零分布进行比较,我们可以计算出P-value:即在零假设下,出现等于或更极端观测结果的概率。如果P-value很小(例如低于预设的显著性水平α),我们就认为零假设不太可能成立,从而拒绝零假设,认为观测结果是统计显著的。 Order by Date Name Attachments image-2 • 66 kB • 207 click 2025年12月16日NULL-pvalue […]
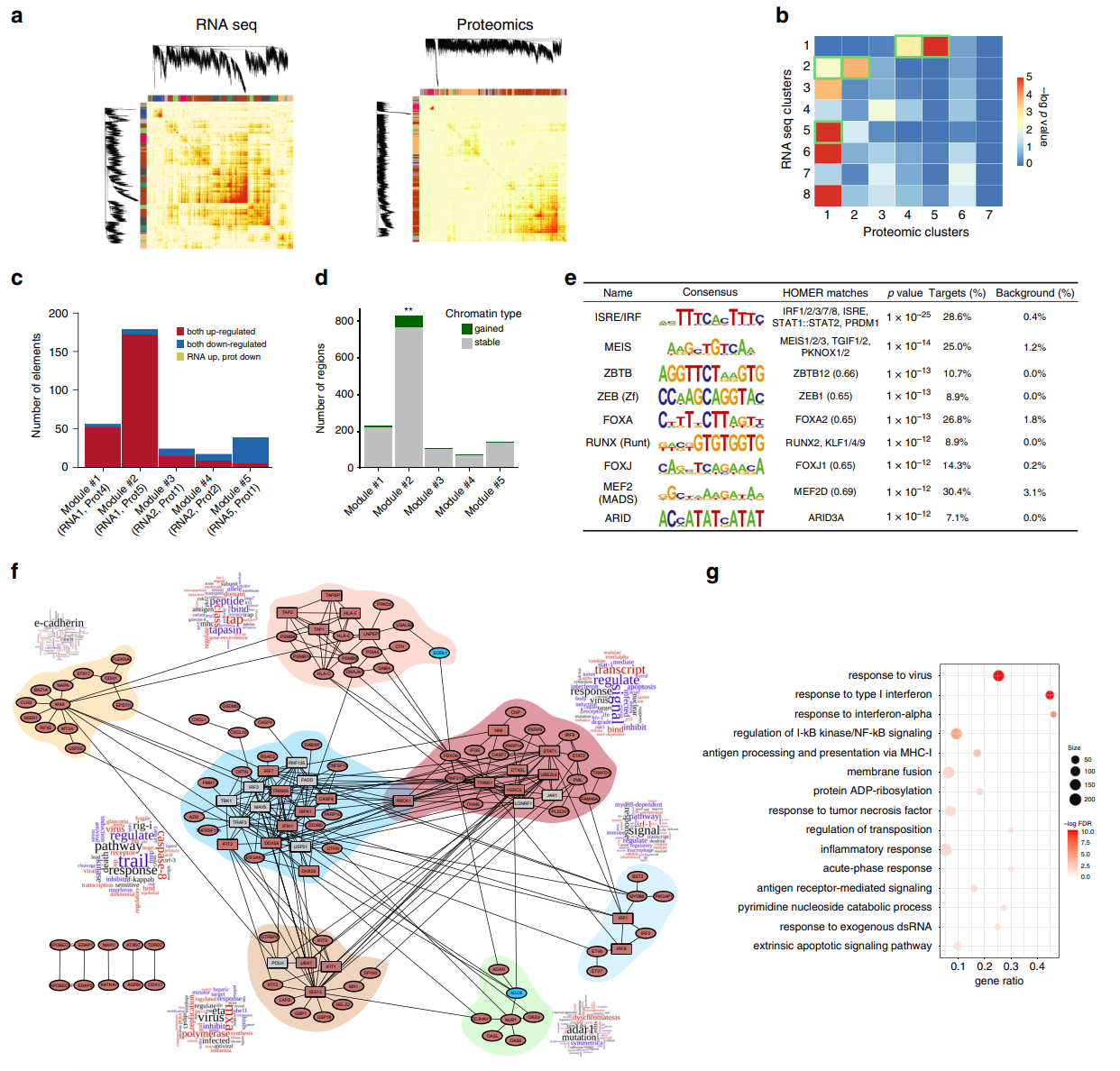
估计阅读时长: 2 分钟Github项目:https://github.com/xieguigang/marker 本程序包是一个基于R语言的综合性机器学习工具集,专门设计用于生物标志物发现和疾病预测模型的构建。该工具整合了多种机器学习算法,提供了从数据预处理、特征选择到模型构建与验证的完整工作流程,特别适用于代谢组学、基因组学等高维生物数据的分析研究。在这个程序包中,主要是通过marker函数来封装了从数据与处理到模型建立的每一个步骤,主要将程序包划分为了以下的工作步骤模块: 数据加载和预处理 初始可视化(PCA图)和统计分析(线性模型、描述性统计) 特征选择(如果未提供预选特征,则使用LASSO、随机森林和SVM-RFE三种方法) 数据分割为训练集和测试集 模型集成训练(逻辑回归、XGBoost、随机森林) 结果可视化(ROC曲线、特征重要性、SHAP分析等) 大家在这里可以通过下面的技术路线图来了解在所编写的程序包中所涉及到的分析内容与步骤: 所主要涉及到的模型算法原理 机器学习方法 数学原理 使用场景 应用 LASSO回归 LASSO(Least […]
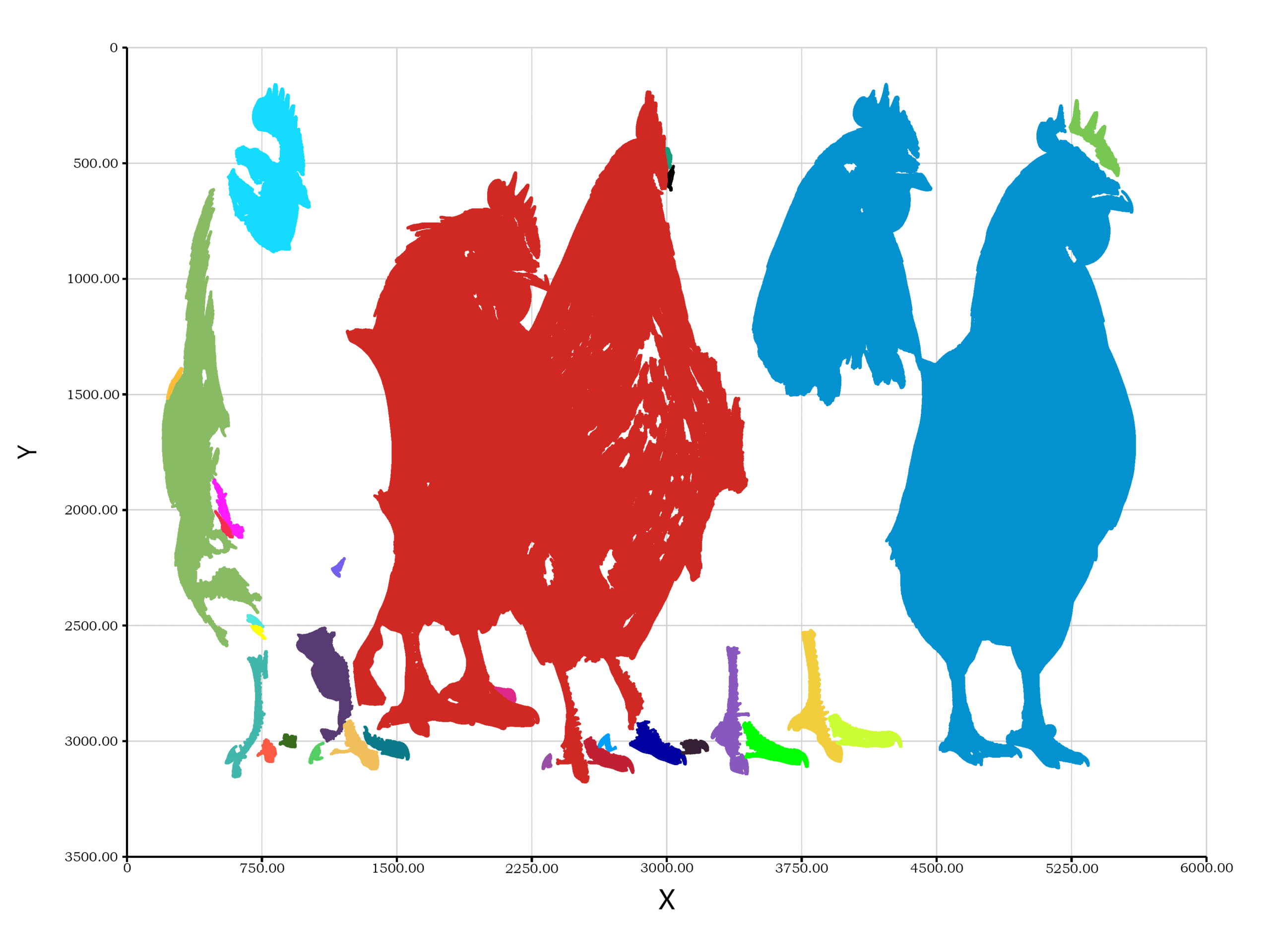
估计阅读时长: 2 分钟Connected Component Labeling(连通组件标记算法)主要用于识别并标记二值图像中相互连接的像素区域(即连通区域)。 imports "geometry2D" from "graphics"; imports "machineVision" from "signalKit"; let raw = readImage("—Pngtree—five chickens […]

估计阅读时长: 7 分钟Boids算法(也称鸟群/鱼群算法)是Craig Reynolds于1986年提出的群体行为模拟模型,通过三条局部规则模拟鸟类、鱼群等生物群体的自组织运动。在Boids算法中,整个过程通过个体(称为“boid”)的局部交互实现全局有序行为,无需中央控制。每条规则计算个体与邻居的相互作用力,最终合力决定运动方向。Boids算法的精髓在于用局部规则涌现全局智能,其简洁性、可扩展性使其成为连接生物行为与工程控制的桥梁。从《蝙蝠侠》的蝙蝠群到无人机编队表演,从游戏生态到交通优化,Boids持续证明:自然界的简单规则,足以驱动复杂系统的有序演化。 Order by Date Name Attachments Boids • 28 MB • 486 click 2025年8月10日Boids • […]

[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?
谢老师,写快点呀,在看着你更新文章呢。
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]