
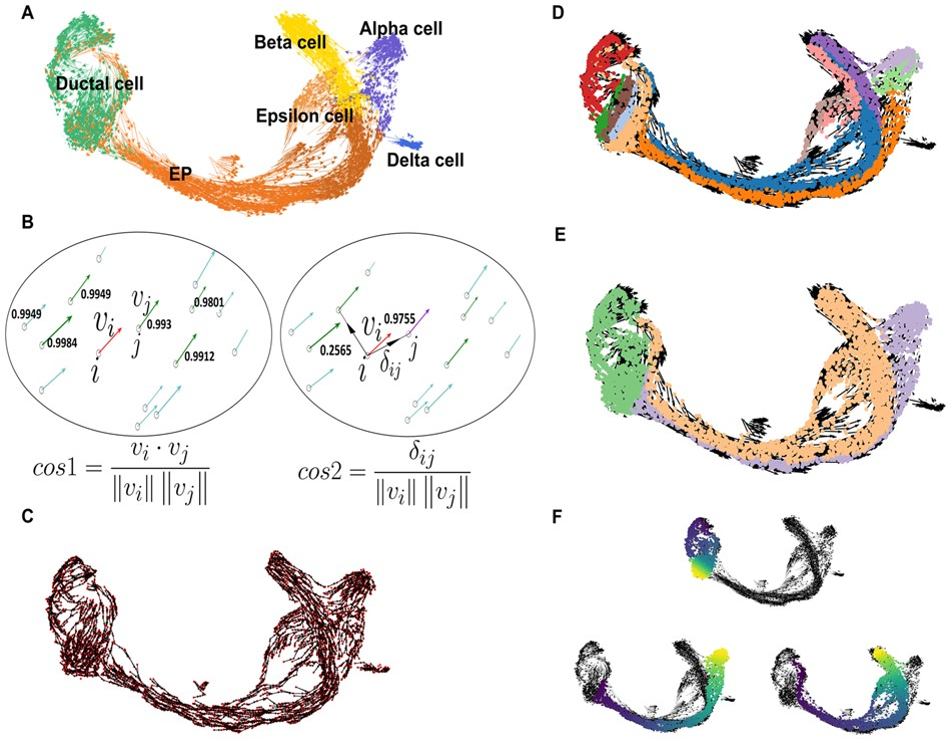
估计阅读时长: 5 分钟单细胞轨迹可以揭示基因调控如何控制细胞命运:大多数细胞状态转变,无论是在发育,重编程或者是疾病异常状态,都以基因表达变化的级联为特征。 Order by Date Name Attachments vec • 722 kB • 680 click 2022年3月17日Slide10 • 14 […]
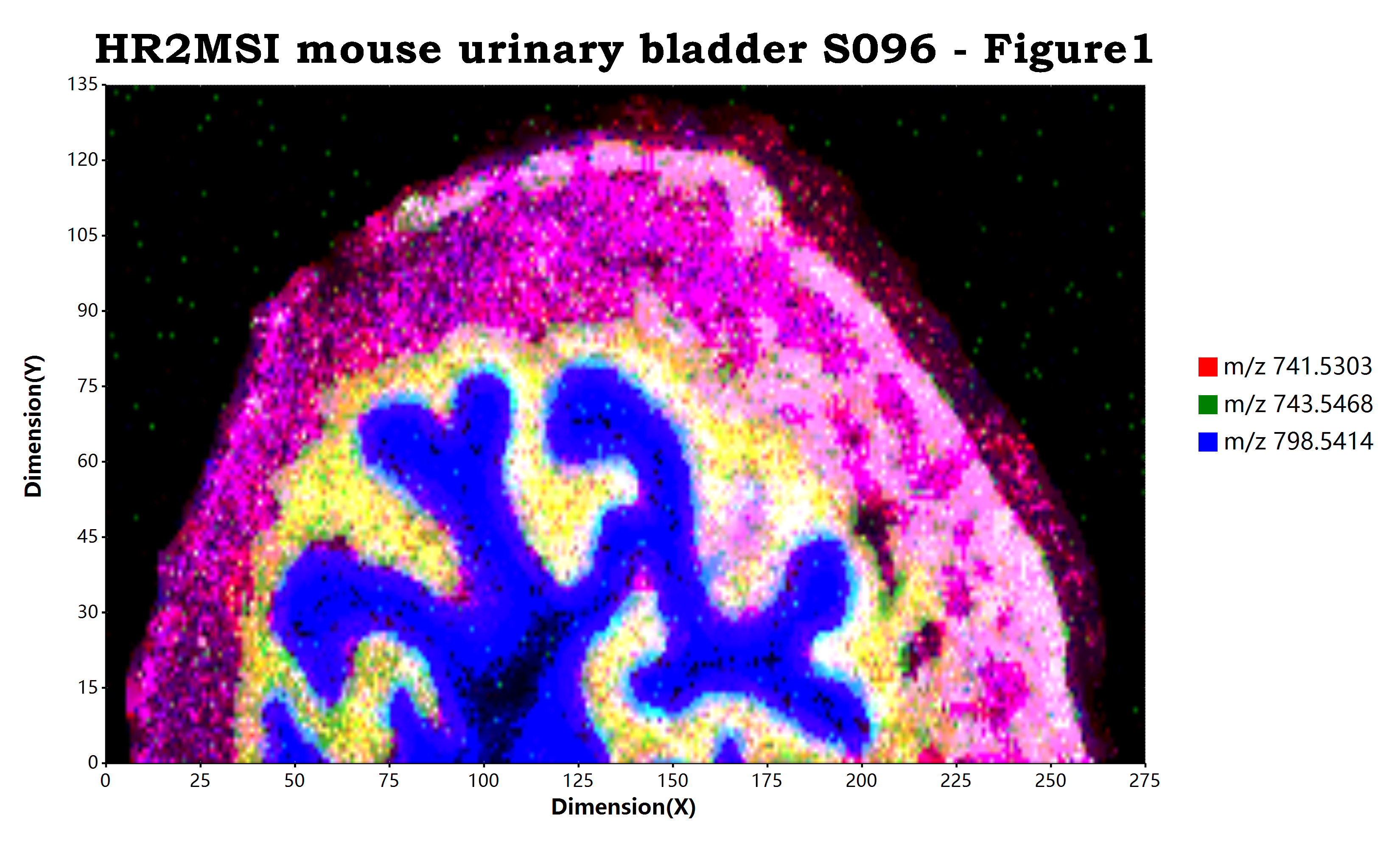
估计阅读时长: 10 分钟https://github.com/xieguigang/ms-imaging Order by Date Name Attachments HR2MSI_mouse_urinary_bladder_S096_RGB • 7 MB • 699 click 2021年11月13日peerj-cs-07-585 • 16 […]
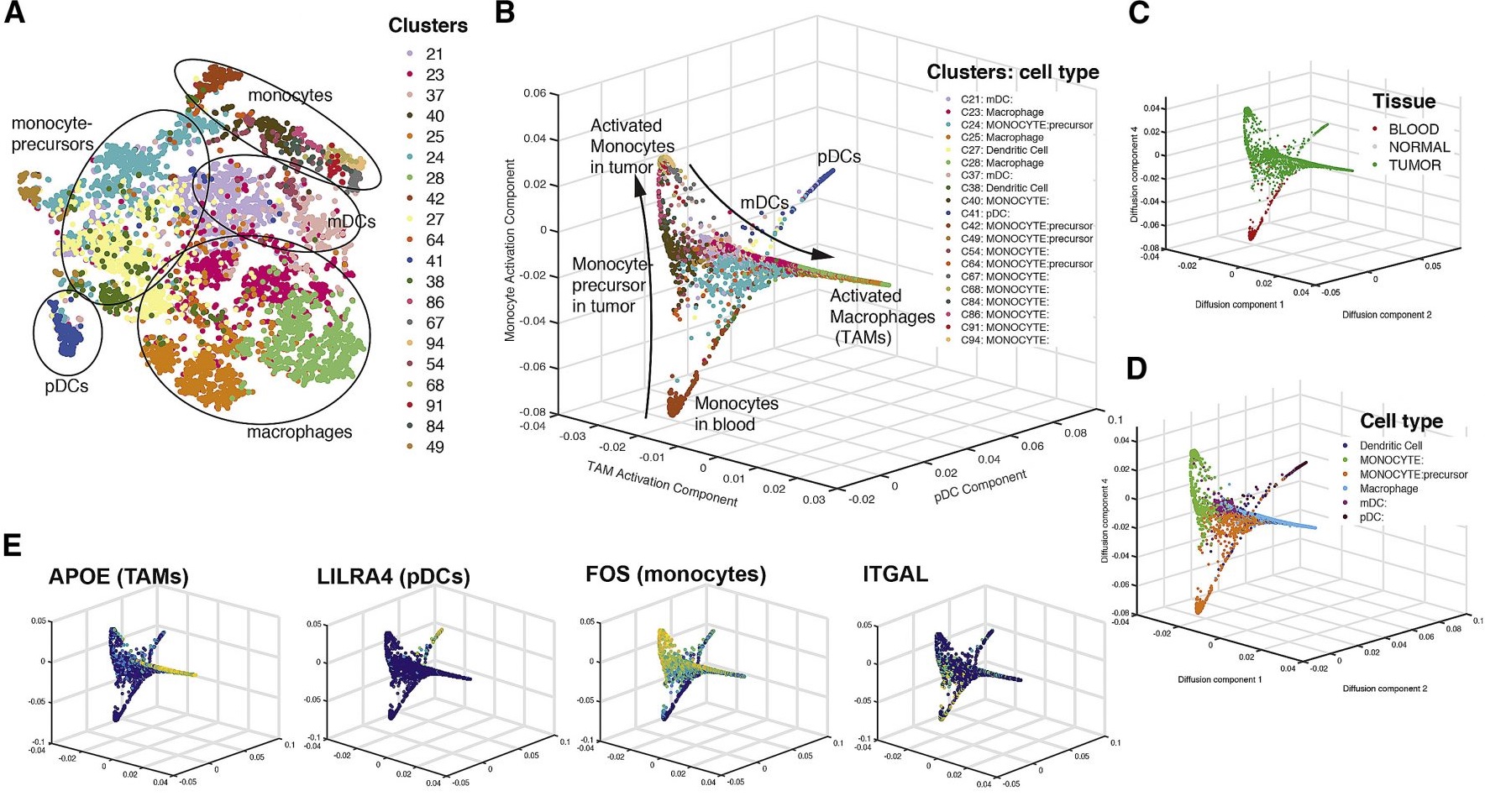
估计阅读时长: 14 分钟https://github.com/rsharp-lang/ggplot 之前在阅读一篇单细胞组学数据分析的文献,觉得在文献之中有一些三维散点图用于展示降维聚类结果的效果非常的好看。于是自己在R#语言之中的ggplot程序包的2D绘图的功能基础之上,进行了三维图形数据可视化功能的开发。 (A) t-SNE map projecting myeloid cells from BC1-8 patients (all tissues). Cells are colored […]
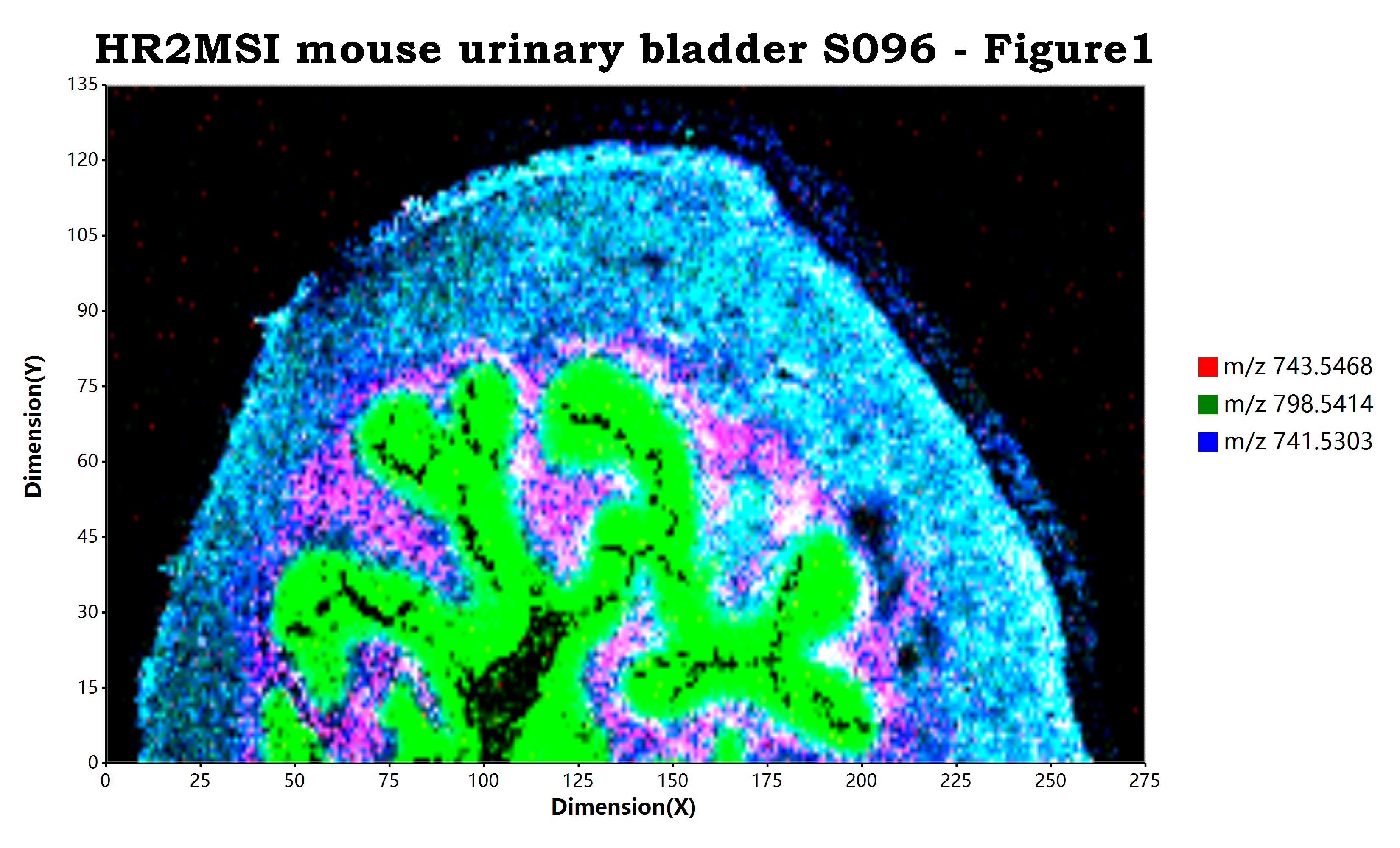
估计阅读时长: 11 分钟https://github.com/xieguigang/ms-imaging Mass spectrometry imaging ( MSI) is a technique used in mass spectrometry to visualize the […]

估计阅读时长: 10 分钟Ascii art是一种使用连续排列的ascii字符进行图形设计的技术。它可以显示在任意的文本框中。ascii art出现于上世纪70年代,最初是当时电脑显示技术不发达时用于显示简单图像的一种娱乐。后来逐渐流行开来,有了专门以此为兴趣的艺术家和研究者。 Order by Date Name Attachments 1537192287563 • 50 kB • 719 click 2021年10月1日ASCII-bilibili […]
估计阅读时长: 15 分钟https://gcmodeller.org 在这篇博客文章之中,我主要是来详细介绍一下是如何从头开始实现Phenograph单细胞分型算法的。在之前的一篇博客文章《【单细胞组学】PhenoGraph单细胞分型》之中,我们介绍了Phenograph算法的简单原理,以及一个在R语言之中所实现的Phenograph算法的程序包Rphenograph。在这里我主要是详细介绍在GCModeller软件之中所实现的VisualBasic语言版本的Phenograph单细胞分型算法。 Attachments Rphenograph • 236 kB • 664 click 2021年9月20日
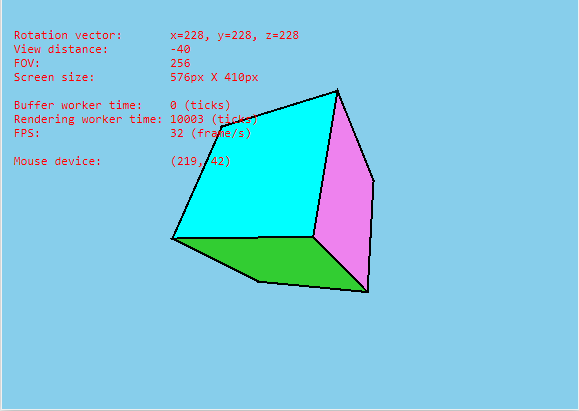
估计阅读时长: 17 分钟https://github.com/xieguigang/sciBASIC/tree/master/gr/Microsoft.VisualBasic.Imaging/Drawing3D 因为大家大多数都是从小接受电子游戏,所以长大了之后能够自己从零开始开发一个完整的3维图形引擎是每一个男程序员的梦想。这个就像玩机械的男人的梦想就是自己从头开始组装一辆汽车。还好这个梦想我在几年前就已经实现了。 Order by Date Name Attachments Cube3D_VB.NET • 4 MB • 746 click 2021年9月19日Cube_screenshot • […]
估计阅读时长: 6 分钟之前在阅读一个使用rust语言编写的contour tracing算法模块的源代码的时候,其中有一个向量的左旋以及右旋的操作。这个操作的具体的含义是和在算法中的轮廓边缘像素的读取方向有关:因为访问方向是一个二维平面的概念,但是在代码中我们只能够使用一个一维的数组的来存储这个二维的信息。所以在这段rust代码之中,作者很巧妙的使用了向量的左旋以及右旋操作来实现一维数组中对二维平面上的方位的访问操作。 Order by Date Name Attachments RotateVector • 30 kB • 683 click 2021年9月16日Full • […]
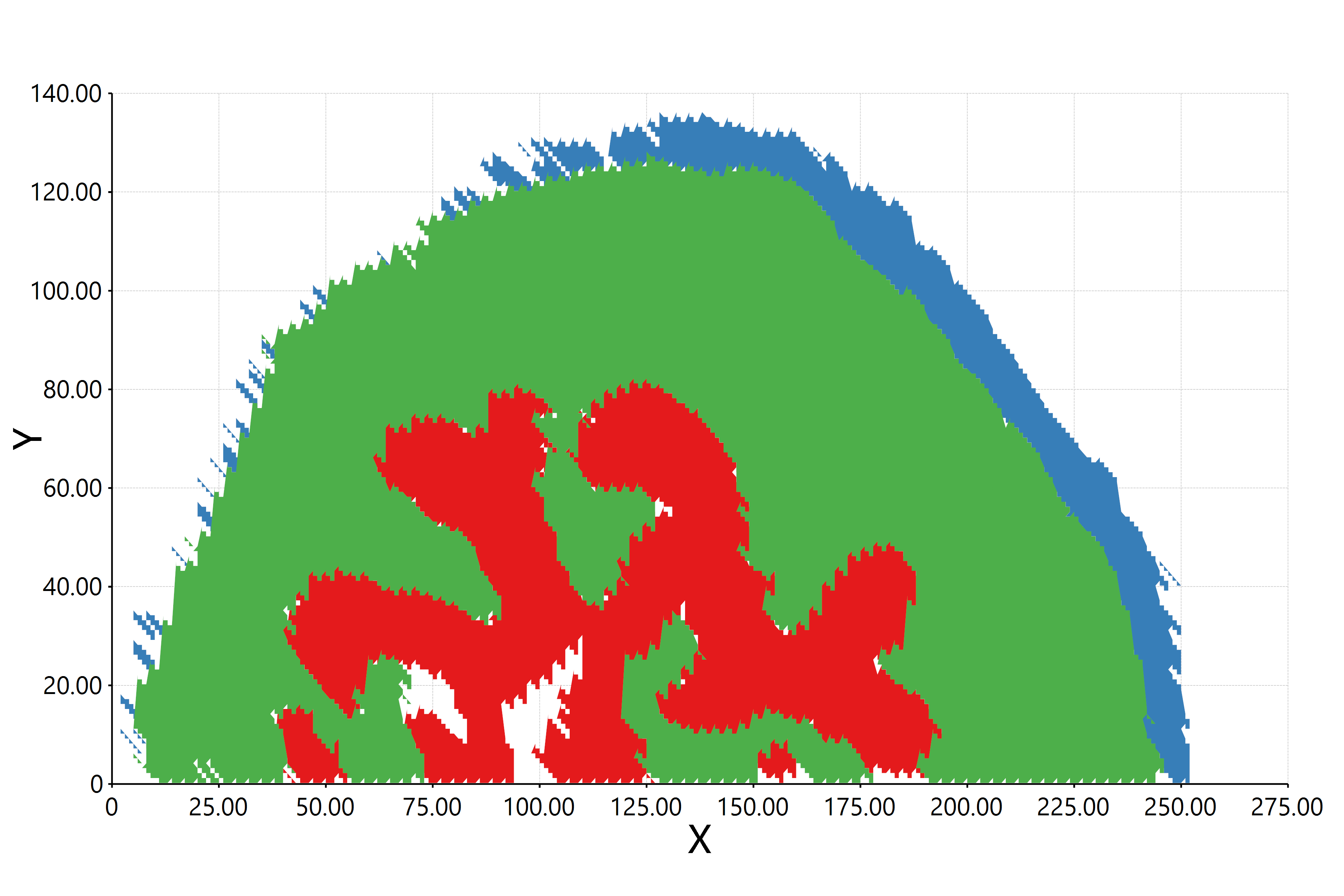
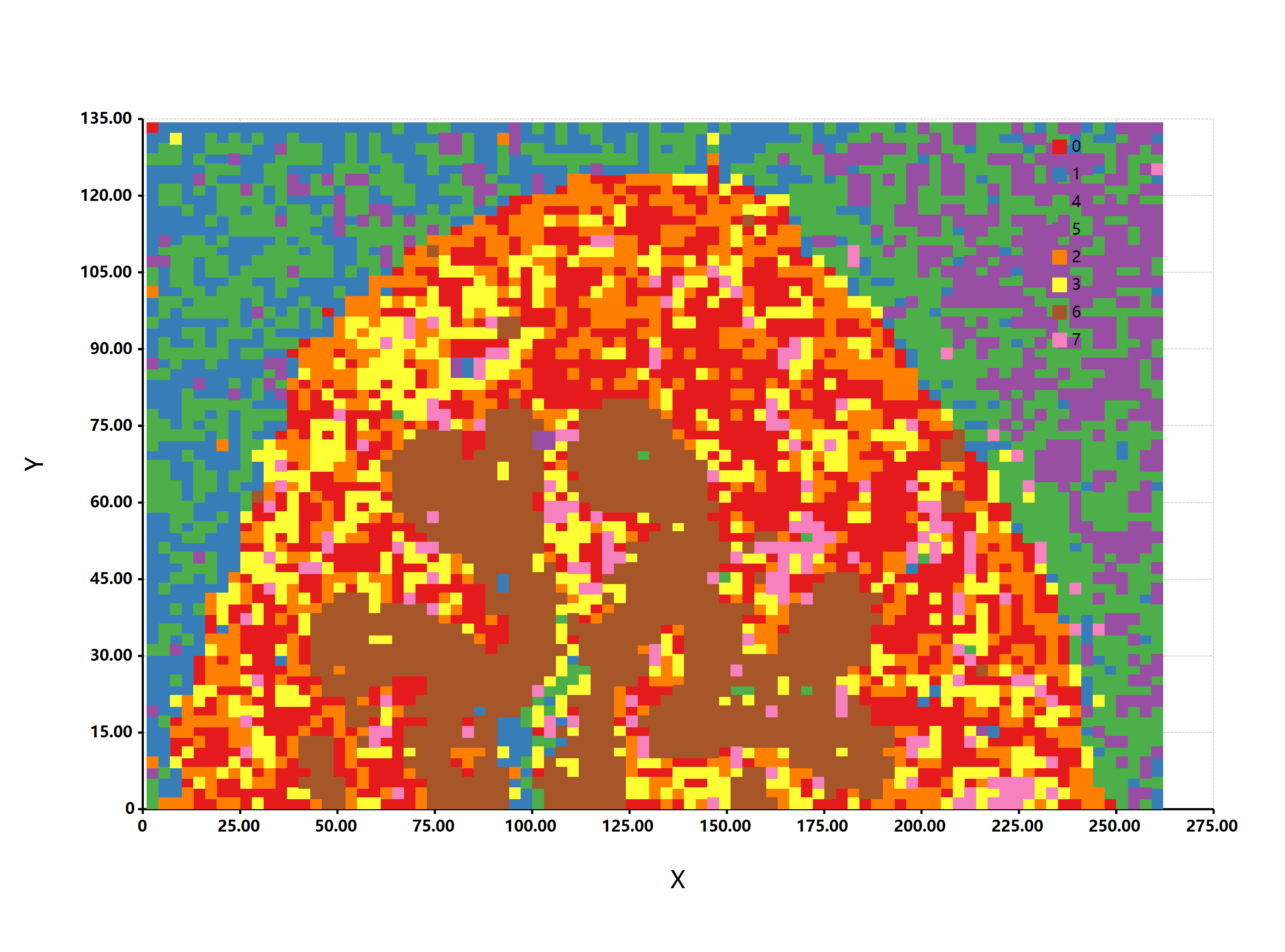
估计阅读时长: 11 分钟https://github.com/xieguigang/sciBASIC 最近在研究实现空间代谢组学中的一些特征区域的自动化划分分割。在得到了特征点集合之后,我们需要根据一些图像处理算法进行特征区域的提取操作。之前,我们尝试过基于绘制等高线图Marching Squares算法的方式来将特征点集合自动转换为特征区域的多边形,实现轮廓扫描获取的功能。但是实现的效果嘛,和实际的区域存在着一些较大的差异。 Order by Date Name Attachments HR2MSI mouse urinary bladder S096 - spatial regions […]

估计阅读时长: 15 分钟https://github.com/xieguigang/sciBASIC 最近在空间代谢组学中的质谱成像应用开发过程中,会需要使用到一些图像处理算法对原始的质谱成像结果图片进行诸如平滑,放大等处理。顺着图像平滑的算法搜索,通过搜索引擎找到了一个年代比较久远的图像处理算法博客文章,将其中的图像算法重新实现了一下,在这里分享给大家。 Order by Date Name Attachments lena • 558 kB • 641 click 2021年9月10日lenalena • […]

[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?
谢老师,写快点呀,在看着你更新文章呢。
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]