
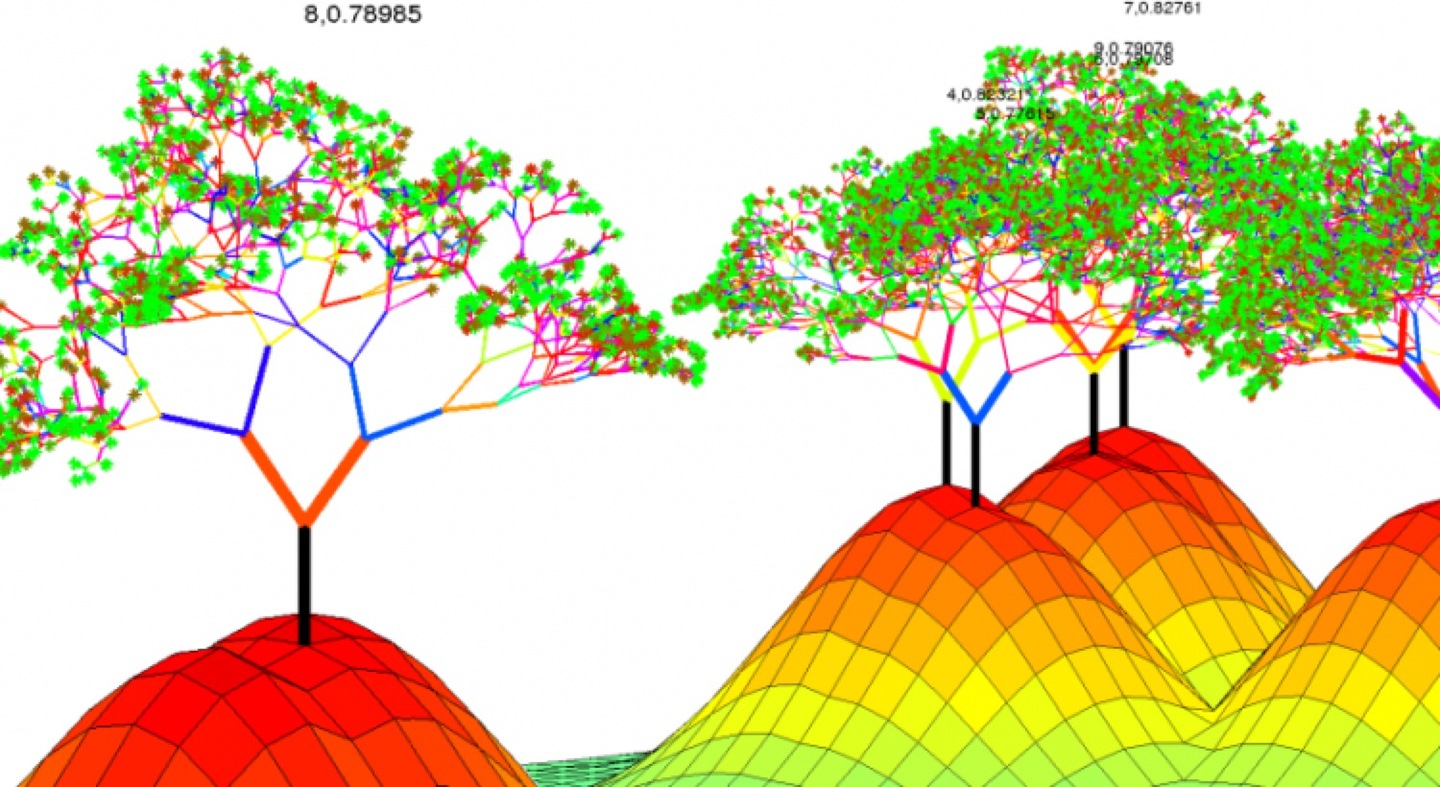
估计阅读时长: 9 分钟https://github.com/xieguigang/sciBASIC 在实际应用的机器学习方法里,GradientTree Boosting (GBDT)是一个在很多应用里都很出彩的技术。XGBoost是一套提升树可扩展的机器学习系统。XGBoost全名叫(eXtreme Gradient Boosting)极端梯度提升。它是大规模并行boosted tree的工具,XGBoost 所应用的算法就是 GBDT(gradient boosting decision tree)的改进,既可以用于分类也可以用于回归问题中。 Order by Date Name […]
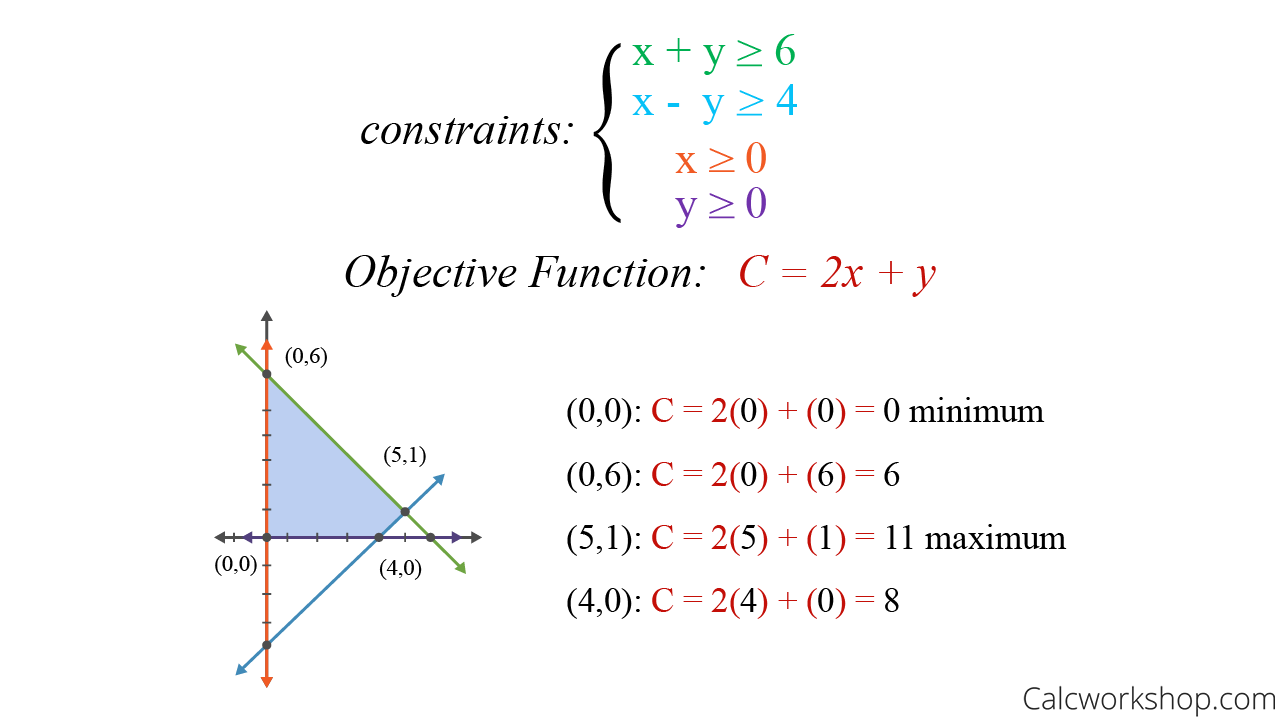
估计阅读时长: 30 分钟https://github.com/xieguigang/sciBASIC/ 线性规划(Linear programming,简称LP)方法起源于20世纪40年代,由美国数学家乔治·丹齐格(George Dantzig)提出,并设计了著名的“单纯形法”。这种优化算法是运筹学中研究较早、发展较快、应用广泛、方法较成熟的一个重要分支,它是辅助人们进行科学管理的一种数学方法。研究线性约束条件下线性目标函数的极值问题的数学理论和方法。通俗点的来讲,就是我们基于这一种数学优化技术,用于在一组线性约束条件下,求解线性目标函数的最大值或最小值(就是在“有限资源”和“一定规则”下,找到“最佳方案”的一种方法)。 Order by Date Name Attachments linear-programming-example • 22 kB • 770 click […]
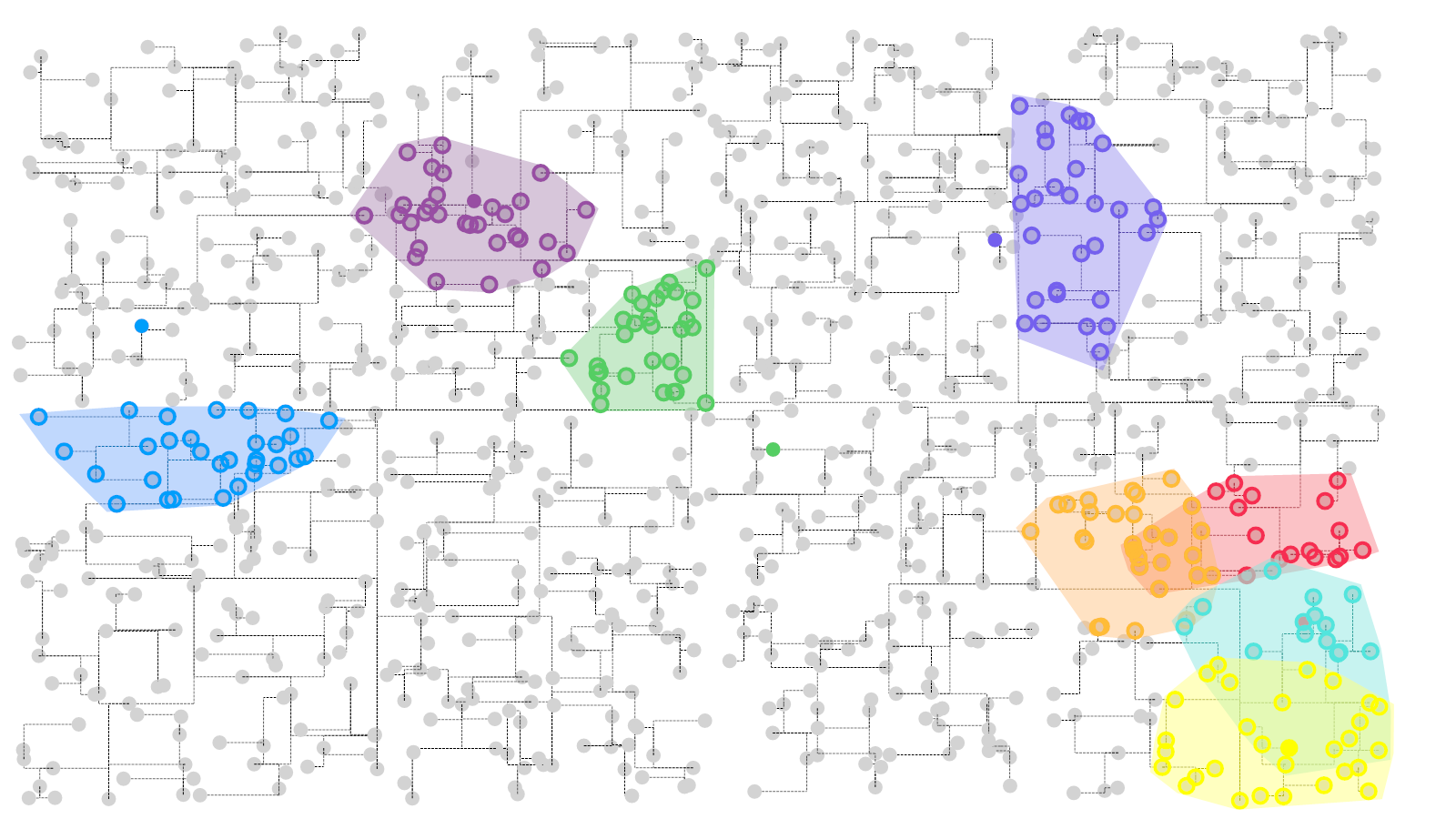
估计阅读时长: 8 分钟https://github.com/xieguigang/sciBASIC 在进行无监督聚类分析的方法之中,我们在算法代码之中一般会遇到求解与某一个样本数据点最相似的数据点的计算过程。对于这个计算过程,一般而言我们是基于欧几里得距离来完成的。 Order by Date Name Attachments Visual a KDtree Search • 274 kB • 748 […]
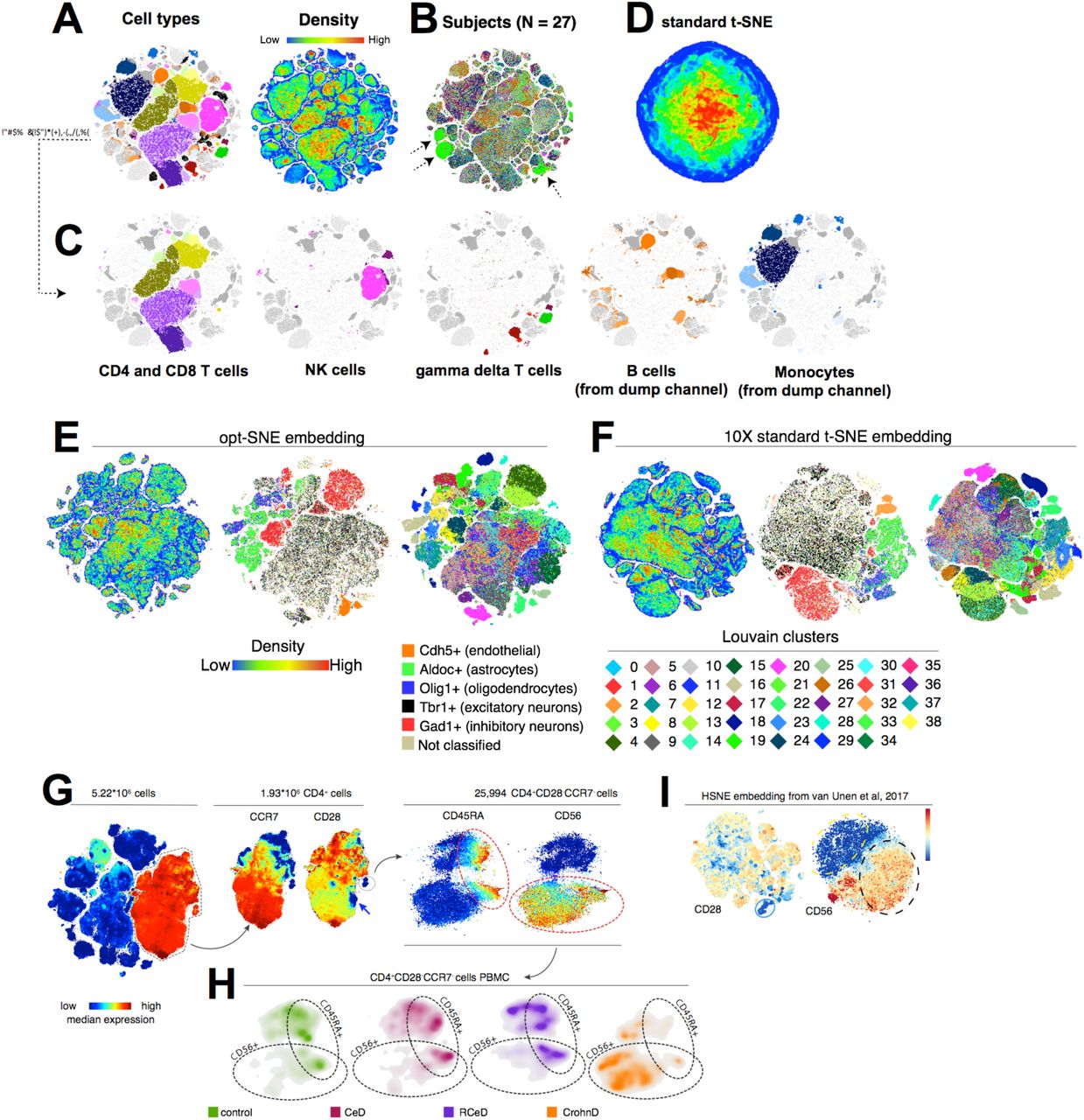
估计阅读时长: 11 分钟PhenoGraph提供了与UMAP类似的算法过程进行单细胞组学数据的细胞分型处理操作。与UMAP方法相比,PhenoGraph并不会产生数据降维效果,仅仅产生数据点Cluster信息。如果需要将数据进行可视化,还需要借助于t-SNE算法将PhenoGraph的分型结果数据投影到一个二维平面上完成。 Order by Date Name Attachments Phenograph-image4 • 200 kB • 687 click 2021年8月9日Automated Optimal Parameters […]
估计阅读时长: 8 分钟https://github.com/xieguigang/sciBASIC/tree/master/Data_science/Mathematica/SignalProcessing 进行峰识别是在代谢组学原始数据分析之中进行定量分析的很重要的一环。在代谢组学之中,定量分析分为靶向定量,以及非靶向定量计算这两大部分。 Order by Date Name Attachments Figure12.36 • 50 kB • 716 click 2021年7月10日view_signal • […]
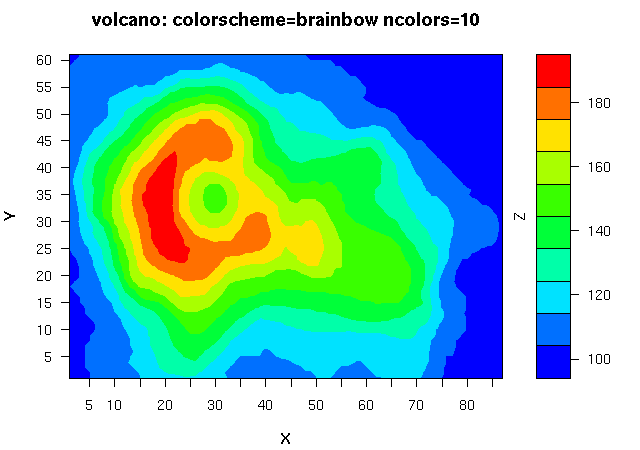
估计阅读时长: 16 分钟https://github.com/xieguigang/sciBASIC 等高线指的是地形图上高程相等的相邻各点所连成的闭合曲线。把地面上海拔高度相同的点连成的闭合曲线,并垂直投影到一个水平面上,并按比例缩绘在图纸上,就得到等高线。 Order by Date Name Attachments 1_Contour • 487 kB • 858 click 2021年6月30日Ms1Contour • […]
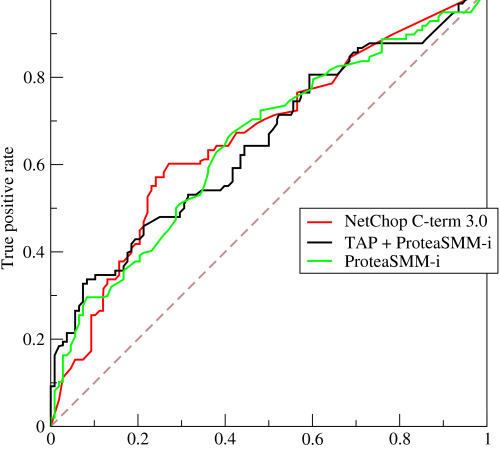
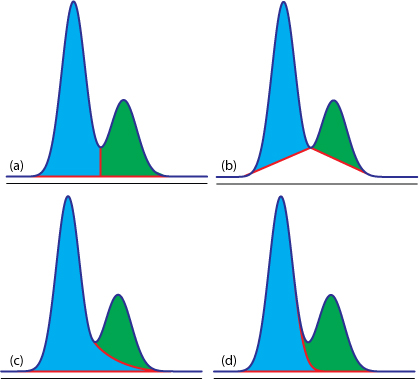
估计阅读时长: 8 分钟https://github.com/rsharp-lang/R-sharp 对于0,1两类分类问题,一些分类器得到的结果往往不是0,1这样的标签。如神经网络得到诸如0.5,0.8这样的分类结果。这时,我们人为取一个阈值,比如0.4,那么小于0.4的归为0类,大于等于0.4的归为1类,可以得到一个分类结果。同样,这个阈值我们可以取0.1或0.2等等。 Order by Date Name Attachments ROC • 221 kB • 762 click 2021年6月28日Roccurves • […]
估计阅读时长: 23 分钟https://github.com/rsharp-lang/R-sharp 降维是将数据由高维约减到低维的过程而用来揭示数据的本质低维结构。它作为克服“维数灾难”的途径在这些相关领域中扮演着重要的角色。在过去的几十年里,有大量的降维方法被不断地提出并被深入研究,其中常用的包括传统的降维算法如PCA和MDS;流形学习算法如UMAP、t-SNE、ISOMAP、LE以及LTSA等。 Order by Date Name Attachments MNIST-LabelledVectorArray-60000x100 • 230 kB • 856 click 2021年6月27日MNIST-LabelledVectorArray-60000x100Euclidean_Distance • […]
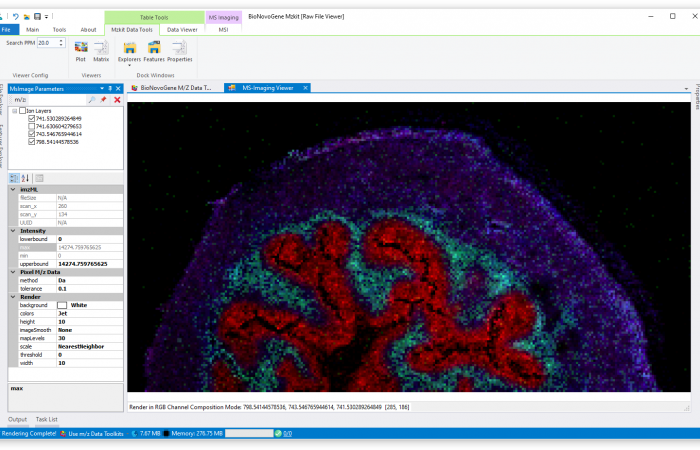
估计阅读时长: 3 分钟http://mzkit.org/ 质谱成像是以质谱技术为基础的成像方法,该方法通过质谱直接扫描生物样品成像,可以在同一张组织切片或组织芯片上同时分析数百种分子的空间分布特征。 Order by Date Name Attachments HR2MSI mouse urinary bladder S096 - optical image • […]

估计阅读时长: 13 分钟https://github.com/xieguigang/voyager-1 旅行者一号是一艘由NASA在1977年9月5日发射的宇宙飞船,其只比旅行者2号晚16天发射。旅行者一号除了担负着研究我们的太阳系的任务之外,在这艘飞船之上还搭载着一张我们尝试对外界介绍我们的文明的一张名片为“地球之音”的铜质镀金激光唱片,这张金唱片承载着人类与宇宙星系沟通的使命。 Order by Date Name Attachments 1080px-The_Sounds_of_Earth_Record_Cover_-_GPN-2000-001978 • 330 kB • 787 click 2021年6月18日scripting • […]

[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
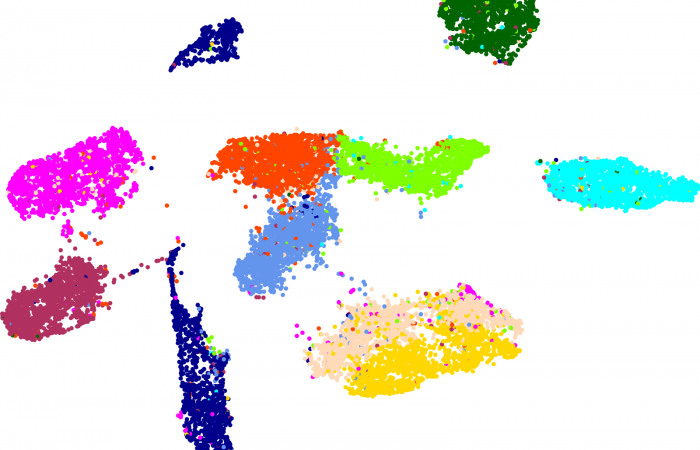
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?
谢老师,写快点呀,在看着你更新文章呢。
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]