
估计阅读时长: 5 分钟基因组 为了能够实现上面所描述的这种基于EC Number的不同层级的加权嵌入,我们在原来的基因组嵌入工具上添加了一个hierarchical选项,用于支持切换为层级嵌入的操作: Imports Microsoft.VisualBasic.Data.Framework Imports Microsoft.VisualBasic.Data.NLP Imports SMRUCC.genomics.Interops.NCBI.Extensions.Pipeline Public Class GenomeMetabolicEmbedding ReadOnly vec As New […]
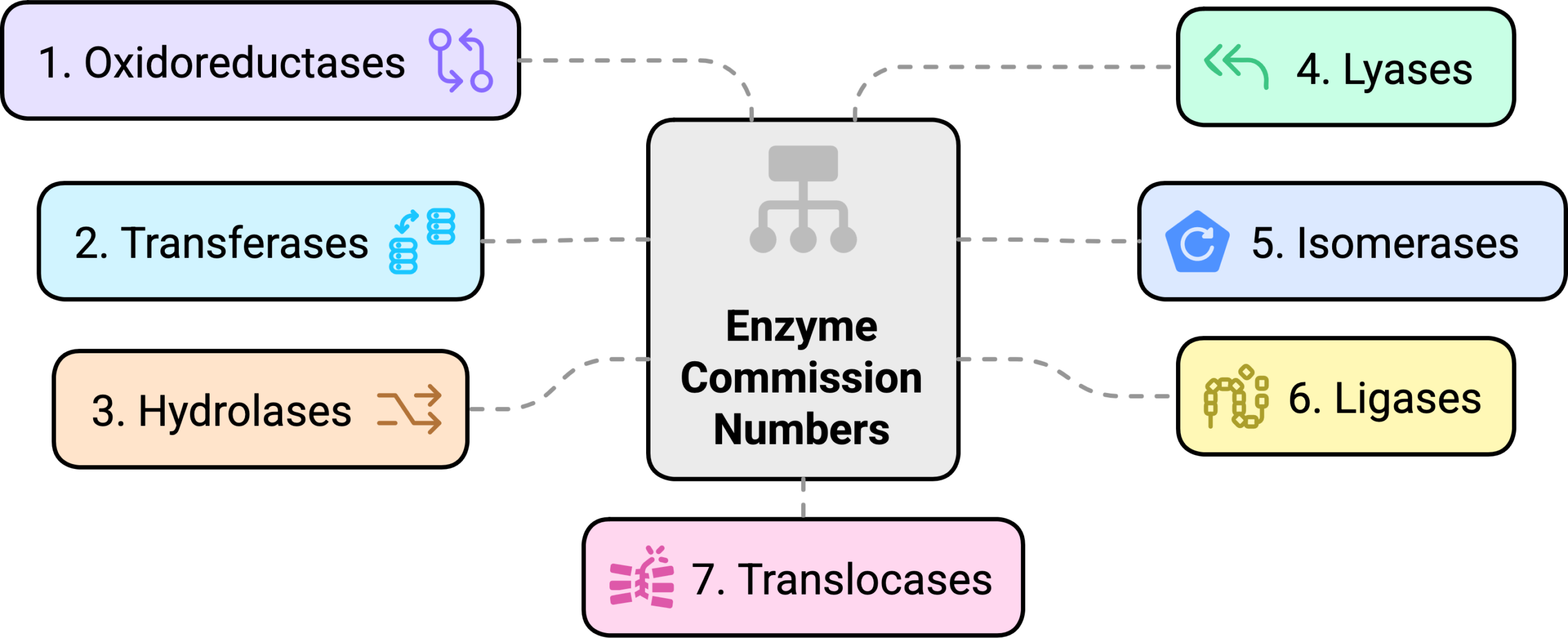
估计阅读时长: 14 分钟酶的EC number(Enzyme Commission number)是国际生物化学与分子生物学学会(IUBMB)酶学委员会制定的酶分类与命名体系的核心标识符,自1961年首次发布以来,已成为酶学研究、数据库管理和生物技术应用的全球标准。这套由四位数字所构成的的层级编码系统不仅解决了早期酶命名混乱的问题,还通过系统化分类揭示了酶催化功能的内在逻辑,为酶学研究提供了统一的框架。 Order by Date Name Attachments Enzyme_Commission_Numbers-visual_selection • 70 kB • 76 click […]

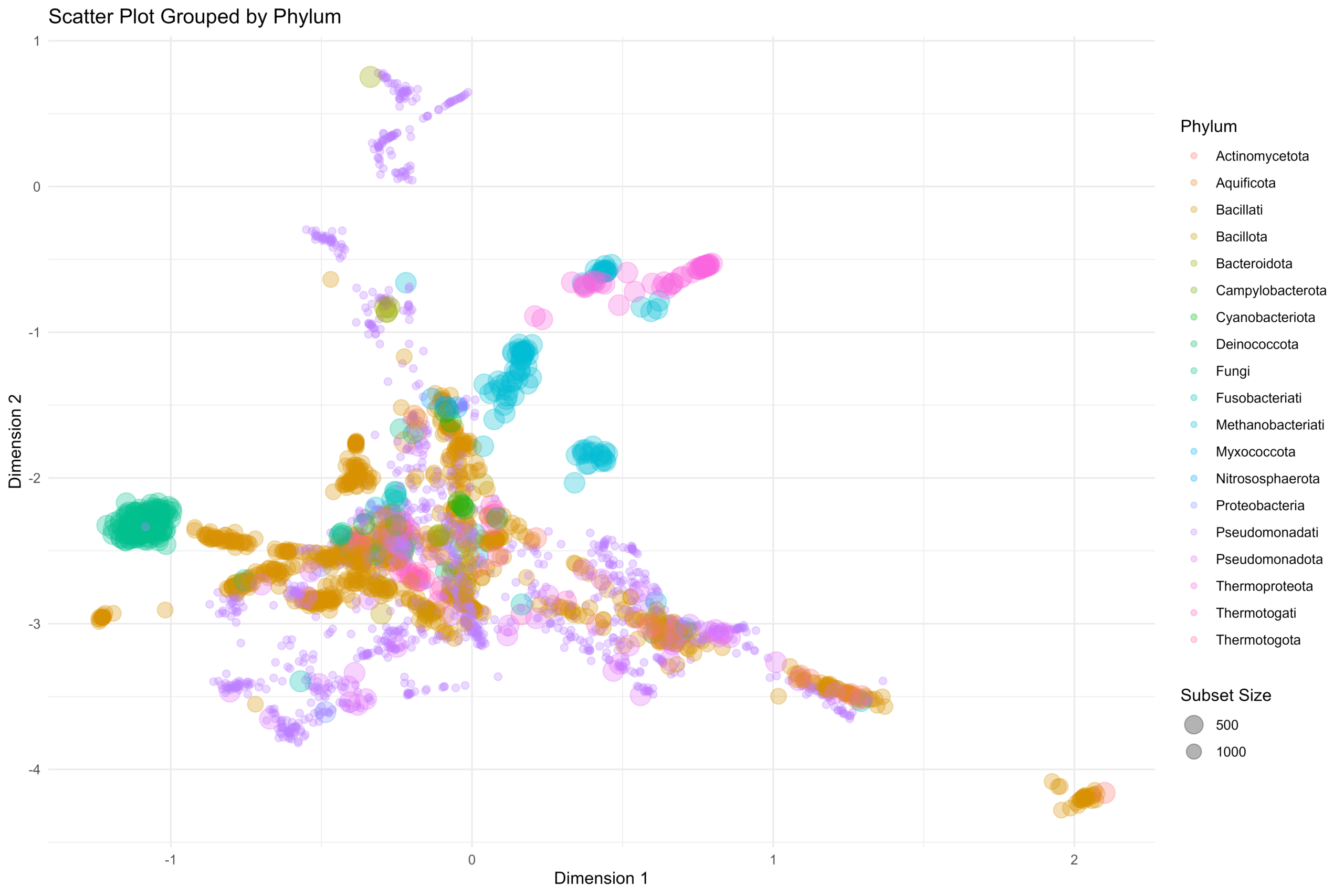
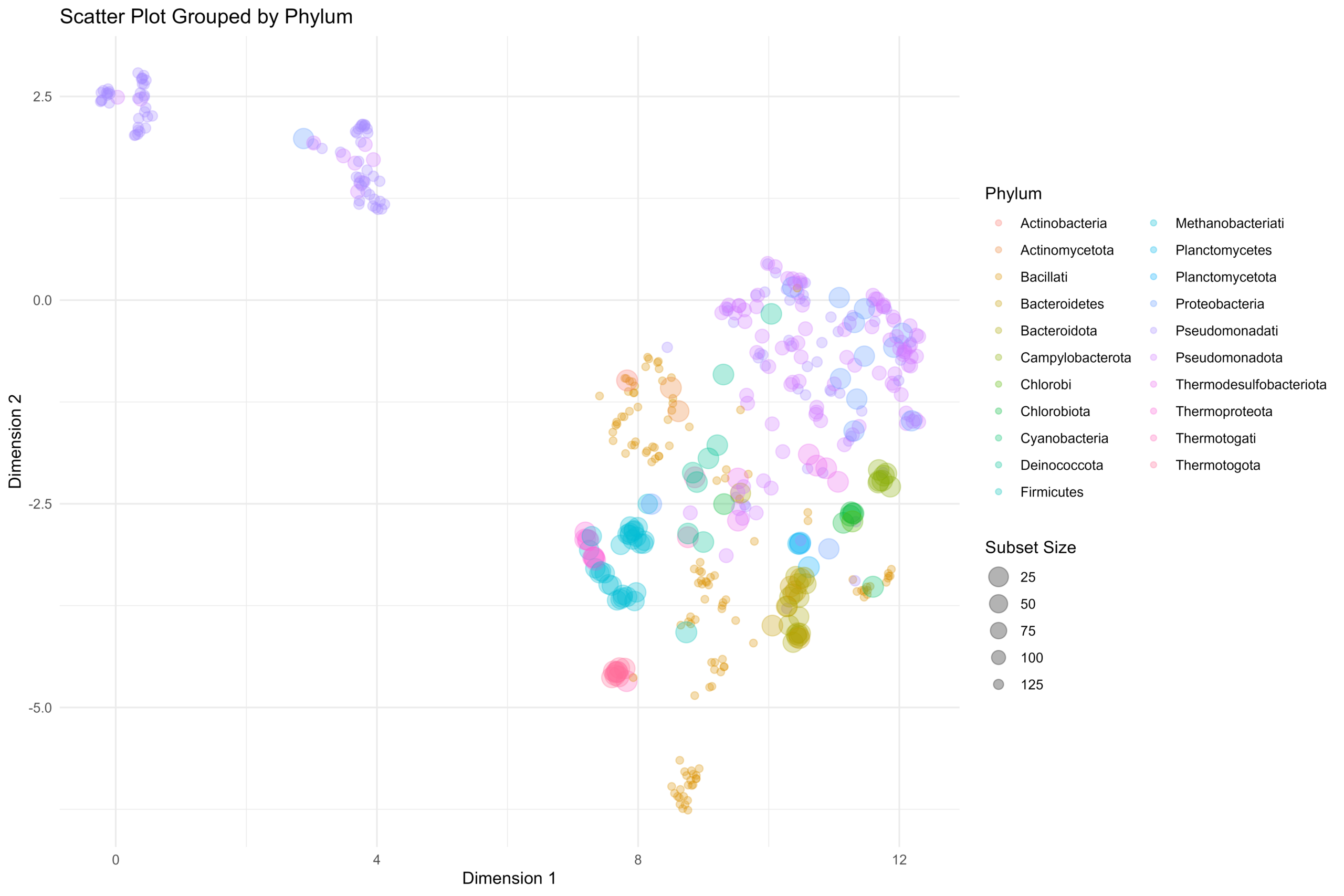
估计阅读时长: 18 分钟在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。除了针对降维后的数据进行散点图可视化,我们还可以直接针对向量化嵌入后的原始嵌入矩阵进行聚类,完成聚类结果的可视化。在这里我们主要是基于嵌入的原始结果进行二叉树聚类可视化。 Order by Date Name Attachments community_metabolic_tree • 220 kB • 77 click 2026年2月15日community-local • […]
估计阅读时长: 20 分钟最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 Order by Date Name Attachments diamond-output • 42 kB • 84 click 2026年2月15日diamond-table-size […]
估计阅读时长: < 1 分钟UPGMA(Unweighted Pair Group Method with Arithmetic Mean,非加权配对组平均法)是一种经典的基于距离矩阵构建系统发育树的聚类算法。其核心思想是假设进化速率恒定(分子钟假说),通过迭代合并距离最近的两个类群(或序列)来构建树。UPGMA算法具有原理简单,计算速度快,易于理解和实现。对于符合分子钟假说(即所有分支进化速率相同)的数据,能给出正确的拓扑结构这些优点。但是其“进化速率恒定”的假设在现实中常常不成立。如果数据存在明显的速率差异(即存在长枝),UPGMA可能会构建出错误的树(拓扑结构错误)。因此,它更适用于进化速率相对均匀的近缘物种或基因的比较。
估计阅读时长: 17 分钟EC Number是国际酶学委员会(IUBMB)制定的一套酶分类编号体系,EC Number采用层级分类法,由4个数字组成,分别代表酶的大类、亚类、亚亚类和序号。例如,“EC 1.1.1.37”中,第一个“1”表示氧化还原酶大类;第二个“1”表示作用于CH-OH基团;第三个“1”表示以NAD+或NADP+为受体的酶;第四个“37”表示特定酶苹果酸脱氢酶。这种层次结构意味着EC编号蕴含了丰富的功能信息,包括酶催化的反应类型和底物/机制。将EC Number嵌入为向量,有助于我们利用机器学习模型进行功能预测、相似性分析等。 Order by Date Name Attachments Capture • 14 kB • 105 […]
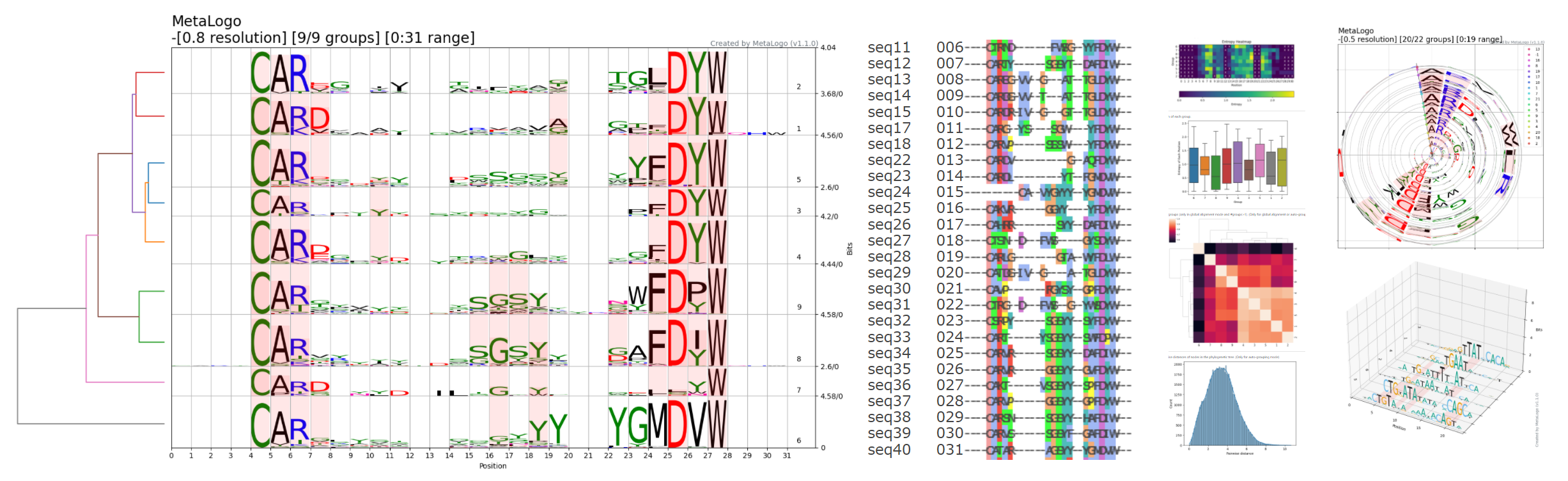
估计阅读时长: 22 分钟MSA(多序列比对)在生物信息学中的核心目标是:通过把多条同源序列“对齐”,来突出它们之间的相似与差异,从而帮助我们:识别保守区/功能位点、推断进化关系(系统发生)、预测或解释蛋白质/核酸结构、发现共进化与功能模块,以及为后续分析(如模体搜索、结构建模、从头设计等)提供基础。基于多序列比对分析,我们可以通过这种算法,把一堆表面看上去“乱糟糟”的序列,整理成一个可以“逐位点比较”的框架。基于我们所得到的这个框架基础,我们可以进行下游的后续分析,例如: 识别哪些部分是“不能动”的(功能/结构核心); 推断它们是如何“进化而来”的(系统发生); 推测它们在空间中“长什么样”(结构预测与建模); 找出哪些部分“一起变化”(共进化与功能耦合); 并把这些信息封装成模型(HMM、profile)用于大规模搜索与注释。 Order by Date Name Attachments MSA • 174 kB • […]
估计阅读时长: 16 分钟KEGG 里面目前并没有“现成的每个 KO 一条代表性序列 FASTA”这种官方序列数据库,假若我们需要基于KEGG数据库中的KO信息的注释,那我们一般会需要自己从 KEGG GENES 里面把每个 KO 对应的基因/蛋白序列抓出来,再按 KO 编号组织成 fasta 集合构建出对应的数据库。基于所建立好的KEGG基因序列数据库,我们就可以实现下面的一些基因注释工作: 在全基因组规模代谢网络重建工作中,进行我们的目标基因组中的代谢网络中的酶节点的直系同源推断,从而将我们的目标基因组中的基因映射到具体的KEGG代谢网络上的节点位置,从而重建出代谢网络模型(使用带有KO编号的蛋白序列做比对注释) 假若我们在进行宏基因组的基因丰度的计算,则可以基于所建立的KEGG基因序列数据库作为参考库,进行宏基因组测序数据中的KO基因丰度的计算(使用带有KO编号的基因序列做比对注释) […]
估计阅读时长: < 1 分钟环境中的微生物往往以复杂群落的形式存在,不同物种之间通过代谢相互作用形成协同或竞争关系,共同完成生物地球化学循环、维持生态系统功能。近年来,随着高通量基因组测序技术的发展,研究者可以从环境样本中获取海量微生物基因组数据,为构建基因组尺度代谢模型(Genome-scale metabolic models, GEMs)提供了基础。GEMs将微生物的全基因组注释与生化反应网络相结合,可以用于模拟微生物在特定环境条件下的代谢能力,预测其生长和代谢产物。在单菌株层面,GEMs已被广泛用于解析微生物对环境变化的代谢适应机制、指导代谢工程设计以及预测药物靶点等。在群落层面,通过将多个GEMs耦合,可以研究微生物之间的相互作用,例如通过代谢物交换实现的协同或竞争关系。 Attachments The-taxonomic-composition-of-various-type-samples-and-the-results-of-neutral-model • 500 kB • 162 click 2026年1月4日
估计阅读时长: 23 分钟Sequence Logo 是一种可视化 DNA 或蛋白质序列保守性的图形表示方法。每个位置(列)上的字母堆叠高度代表该位点的信息含量(以 bits 为单位),而每个字母的高度则与其在该位点出现的频率成正比。高信息量的位置字母堆得高,低信息量的位置则矮甚至接近零。Sequence Logo的绘制遵循信息熵原理,我们可以很直观的通过某一个位置的总高低来了解该处位置的信息含量有多少,高信息量的位置,字母堆的高,一般会出现某一个字符特别高,表明该处非常保守。 位置权重矩阵(Position Weight Matrix, PWM)是描述基因组调控因子结合位点序列模式的核心模型。它通过统计在结合位点序列中每个位置上各核苷酸(或氨基酸)出现的频率,来量化该位置对不同碱基的偏好程度。PWM通常以矩阵形式表示,行对应核苷酸(A、C、G、T/U),列对应序列中的位置,矩阵元素即为该位置该核苷酸相对于背景的权重得分。这一模型简洁且易于计算,因此在转录因子结合位点(TFBS)等调控元件的识别和表征中被广泛采用。 Order by Date Name […]

[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?
谢老师,写快点呀,在看着你更新文章呢。
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]