
估计阅读时长: 11 分钟在进行热图的渲染的时候,我们需要首先将需要进行渲染的数据转换为一个0到1之间的灰度值,然后基于所设定的颜色列表,将灰度值映射为颜色列表的索引号,获取某一个灰度对应的颜色,从而完成对热图的渲染过程。在这个过程中,假若我们是针对热图需要获取得到一个连续的颜色列表,则我们还需要使用插值算法针对基础的关键颜色列表进行插值计算,生成调色板。 Order by Date Name Attachments volcano_ggthemes_Traffic • 17 kB • 455 click 2025年6月12日volcano_ggthemes_excel_Ion_Boardroom • 15 […]

估计阅读时长: 5 分钟https://github.com/xieguigang/scale_colour_genshin 在用R绘图时,颜色设置是美化过程中不可缺少的一步。在实际绘图时,一般不会一一手动寻找合适的颜色,而是通过一些R包、网站提供好的,美观的颜色组合,即调色板(palette),可供使用。在这里介绍一种通过提取图片主题色的方法来为我们自动生成画图所用的颜色板数据。 Order by Date Name Attachments 383807b4 • 132 kB • 672 click 2023年4月8日faruzan • […]
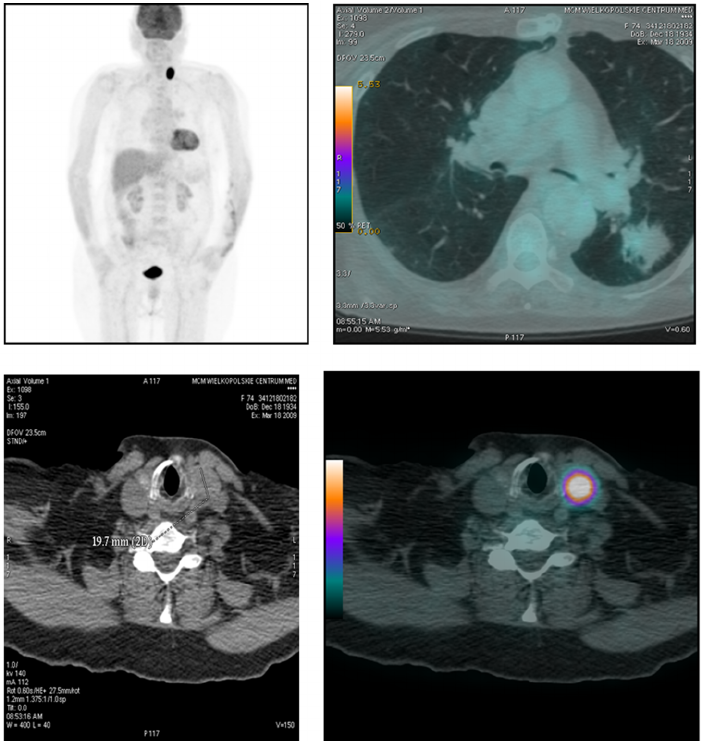
估计阅读时长: 8 分钟https://github.com/rsharp-lang/NRRD NRRD(Nearly Raw Raster Data)是一种用于存储类似于热图成像数据的文件格式。其实我们可以将NRRD看作为类似于bitmap之类的未压缩的原始光栅图像文件。只要我们有对应的解码方式,我们就可以像查看普通图片文件一样查看NRRD文件。 Order by Date Name Attachments raster__238 • 61 kB • 651 […]
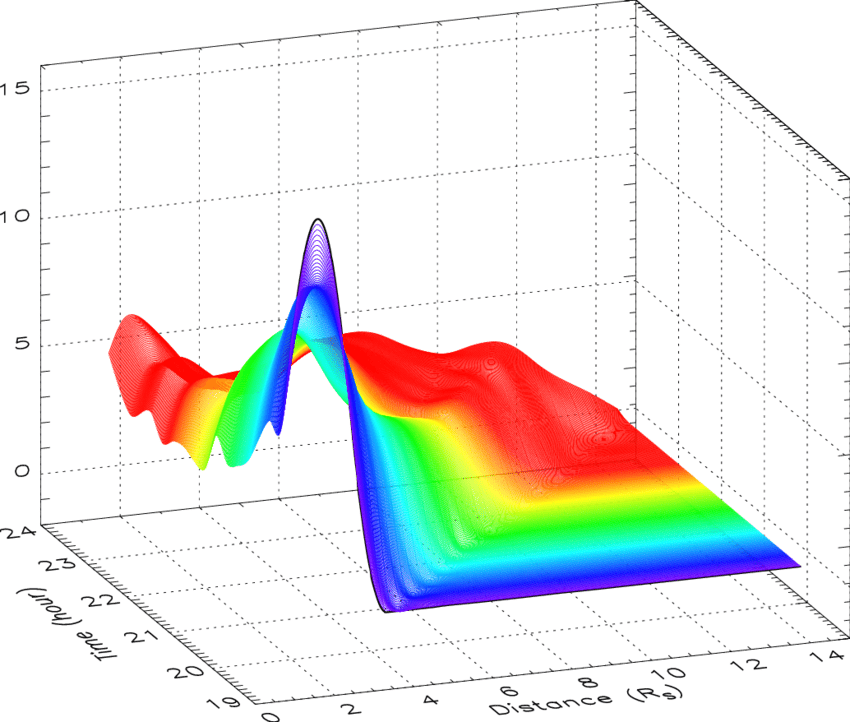
估计阅读时长: 7 分钟热图(Heat Map)是在二维空间中以颜色的形式显示一个现象的绝对量一种数据可视化技术。颜色的变化可能是通过色调或强度,给读者提供明显的视觉提示,说明现象是如何在空间上聚集或变化的。热图有两种完全不同的类别:聚集热图和空间热图。 在聚集热图中,幅度被排列成一个固定单元格大小的矩阵,其行和列是离散的现象和类别,行和列的排序是有意的,而且有些随意,目的是暗示聚集或描绘出通过统计分析发现的聚集。单元格的大小是任意的,但足够大,可以清晰可见。 相比之下,空间热图中某一量级的位置是由该量级在该空间中的位置所决定的,没有单元的概念,现象被认为是连续变化的。 Order by Date Name Attachments 2D-cubic-spline-interpolation-of-mass-profiles-from-1939-to-2354-UT-and-between-16 • 112 kB • 824 click […]
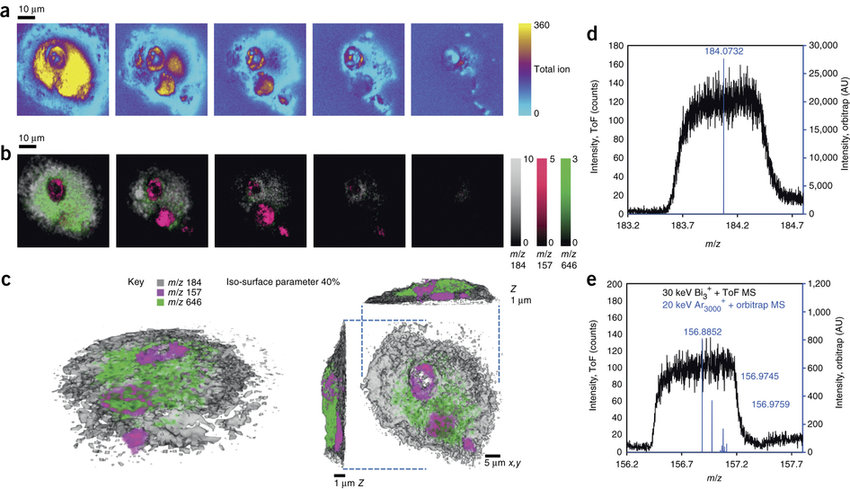
估计阅读时长: 2 分钟在BILIBILI上观看视频:【空间代谢组学】AP-MALDI 质谱成像技术介绍 哈啰,大家好呀,鸽了大半年之后,你们的小姐姐又回来啦。为了更好的制作出质量更高的视频,你们的六神无主鸠小姐姐呀,在这大半年的时间里面一直在努力的学习新技术。经过半年的钻研学习,收获满满。谈到最近几年的热门尖端技术,大家都会谈论到空间转录组和单细胞技术。一般而言,代谢组学的发展要稍微滞后于转录组学研究。最近一年呢,随着空间转录组的热度的降低,空间代谢组的热潮也终于姗姗来迟终于到来了。今天呢,我想要为大家介绍的是在最近几年内出现的,目前比较火热的空间代谢组学研究领域内的质谱成像技术。 Order by Date Name Attachments 3D-MS-imaging-using-dual-beam-and-dual-spectrometer-mode-10-of-single-rat-alveolar • 99 kB • 790 click 2022年5月6日Microsoft […]

[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?
谢老师,写快点呀,在看着你更新文章呢。
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]